Beyond the Nucleus: TDP-43 Sticks Together, For Better or Worse
Quick Links
Similar to a newborn human, nascent RNA transcripts need a lot of coddling before they realize their purpose in life (that is, translation into protein). It takes a village of RNA-binding proteins to accomplish this. They see to the transcripts’ proper splicing, capping, polyadenylation, and export from the nucleus, and they send the finished RNAs off to sometimes far-flung locales such as axonal terminals for translation. When the RNA-binding proteins themselves get mixed up, the cell is in trouble. This goes for FUS (see Part 3 of this series) and for TDP-43. While most TDP-43 works from the neuronal nucleus in healthy people, the protein increasingly acts in the cytoplasm in ALS/FTD. At the 5th RNA Metabolism in Neurological Disease Conference, held November 1-2 in San Diego, researchers exchanged surprising new insights on how the protein gets trapped in the cytoplasm, and hatched plans for new therapeutic targets.
- When TDP-43 dimerizes in the cytoplasm, it accumulates there and goes missing from the nucleus.
- Loss of TDP-43 also means loss of the microtubule binding protein stathmin-2, with dramatic consequences.
- TDP-43 forms amyloid-like “myogranules” with RNA in developing muscle fibers.
Sami Barmada of the University of Michigan, Ann Arbor, connected the dots between TDP-43’s exile in the cytoplasm and neuronal hyperactivity—common phenomena in ALS/FTD. When he stimulated human neurons derived from induced pluripotent stem cells (iPSC) with tetraethylammonium or other activators, he found that this shifted TDP-43 into the cytoplasm. Deploying a collection of N- and C-terminal antibodies against TDP-43, Barmada zeroed in on an unlikely cause. Somehow, neuronal activity led to the appearance of a C-terminally truncated version of TDP-43. It arose from alternative splicing and, strikingly, the truncation introduced a nuclear export signal at the C-terminus. This relegated the shortened protein to the cytoplasm, where it promptly interacted with full-length TDP-43. Barmada proposed that this stunted form of TDP-43 promotes cytoplasmic accumulation of full-length TDP-43. In support of this idea, more full-length TDP-43 piled up in the cytoplasm when Barmada co-expressed the short isoform in neurons.
The findings not only offer a new explanation for how TDP-43 finds itself in the cytoplasm, they could also explain discrepancies in past data depending on whether N- or C-terminal antibodies were used to pinpoint where in the cell TDP-43 was. The more commonly used C-terminal antibodies do not recognize truncated TDP-43, and therefore undercount cytoplasmic localization relative to N-terminal antibodies, which recognize full-length and truncated forms, Barmada said.
What might this short form do, researchers wanted to know? While Barmada has confirmed that it binds RNA, he does not know yet whether it is functional, how it affects full-length TDP-43, or how it interacts with cytoplasmic stress granules.
Previous studies have hinted at oligomerization as a prerequisite for TDP-43’s accumulation and potential entrapment in such granules. To nail down the consequences of this coupling, some investigators use light to force to protein to oligomerize. Christopher Donnelly of the University of Pittsburgh, Pennsylvania, built upon the “optodroplet” system previously developed by Clifford Brangwynne at Princeton University, New Jersey. He fused TDP-43 to cryptochrome 2 (Cry2), a photoreceptor that dimerizes fast when exposed to blue light (Jan 2017 news). In San Diego, Donnelly reported that when cells expressing this Cry2-TDP-43 hybrid were chronically exposed to blue light, the protein left the nucleus and built up in the cytoplasm. There, it formed inclusions that recruited endogenous TDP-43, a hallmark of proteinopathy. Interaction with RNA rendered TDP-43 resistant to this state, indicating that the nucleic acid antagonizes TDP-43’s propensity to form inclusions.
These TDP-43 “opto-inclusions” were more than experimental playthings. Donnelly told the audience that they are toxic, killing cells two days after induction of TDP-43 oligomerization. His team made an “opto-fly” that expresses the Cry2-TDP-43 hybrid, and are starting to see that TDP-43 dimerization triggers motor deficits in the insects. They also showed that treating cells with decoy oligonucleotides composed of TDP-43 binding sequences prevented these oligomerization events and delayed neurotoxicity.
Kazuhide Asakawa of the National Institute of Genetics in Mishima, Japan, expressed a similar Cry2-TDP-43 construct in zebrafish, saying that the see-through minnows make excellent models to study cellular consequences of TDP-43 aggregation. Flashing the fish with pulses of light coaxed TDP-43 out of the nucleus and into the cytoplasm of motor neurons. Echoing Barmada’s findings with short TDP-43, Asakawa reported that these flashes essentially transformed opto-TDP-43 into a dominant-negative protein as it coupled with normal TDP-43 in the cytoplasm. This cytoplasmic mislocalization somehow brought axonal outgrowth to a halt.
Don Cleveland of the University of California, San Diego was curious about Asakawa’s latter result, for reasons that became clear during his own presentation. TDP-43 influences myriad RNA processing steps for numerous transcripts, and its depletion from the nucleus changes the expression level of more than 1,500 genes. How to find the important ones? Cleveland suspects many of the toxic consequences of TDP-43’s nuclear exodus come down to changes in a single transcript. Why? The microtubule binding protein stathmin-2 was the most severely downregulated gene subsequent to TDP-43 deficiency. Cleveland reported that with loss of nuclear TDP-43 somehow came a premature polyadenylation site in the first exon of stathmin-2, resulting in a stunted transcript that produced no functional protein. Notably, he also found stathmin-2 expression to be deficient in neurons generated from ALS/FTD patients with mutations in C9ORF72 or TDP-43, though not SOD1. Neurons from sporadic cases had the defect, too.
Cleveland’s group used a microfluidic model of axonal regeneration to investigate what losing stathmin-2 would do. Motor neurons grew in a compartment that was connected to a neighboring chamber by a segment that allowed axons to project through. When these axons were severed, normal motor neurons rapidly grew new axons. However, antisense oligonucleotides (ASOs) that knocked down either TDP-43 or stathmin-2 completely blocked this regeneration. Notably, restoring stathmin-2 expression alone completely rescued this defect. Cleveland proposed using ASOs targeting the premature polyadenylation site in stathmin-2 as a therapeutic strategy for any form of ALS/FTD marked by TDP-43 pathology.
Researchers peppered Cleveland with questions. Fen Biao Gao, University of Massachusetts Medical School, Worcester, asked whether stathmin-2 expression was abnormal in tissue sections from ALS/FTD patients. Cleveland said he had not looked, and that such an analysis would be limited by the specificity of antibodies; moreover, many vulnerable neurons no longer exist in postmortem sections. Benjamin Wolozin, Boston University, suggested that this result be confirmed using TDP-43 sans its nuclear localization sequence, to be sure that the stathmin-2 loss was due to TDP-43 depletion from the nucleus. When asked what exactly spurs introduction of the polyadenylation site, Cleveland said it was still unclear.
Dieter Edbauer, German Center for Neurodegenerative Diseases, Munich, asked about stathmin-2 in TDP-43 mouse models. Cleveland responded that the polyadenylation site—which is modified from UU to AA in the human stathmin-2 gene—does not exist in mice; they have two Gs in that position instead. Perhaps this is why TDP-43 mouse models do not fully recapitulate disease, Cleveland said. Still, others wondered what explains the ALS/FTD phenotypes that TDP-43 mouse models do have, despite their apparent resistance to premature polyadenylation of stathmin-2.
Despite the unknowns, some researchers were cautiously optimistic about the approach. Gao told Alzforum that stathmin-2 need not explain all TDP-43 toxicity to be a viable target. Others were less convinced. Ke Zhang of Johns Hopkins University in Baltimore challenged Cleveland on his finding, noting that the axonal regeneration phenotype has little to do with neurodegeneration. He believes that without evaluating stathmin-2 loss in animal models more broadly first, pushing stathmin-2 as a therapeutic target is premature.
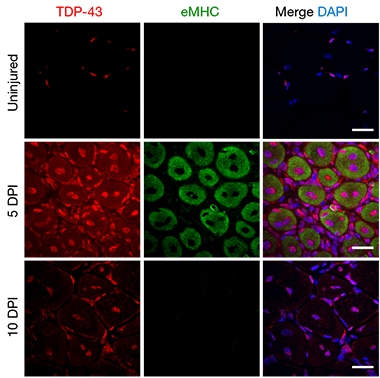
Myogranules to the Rescue?
TDP-43 (red) stays in the nucleus in uninjured myocytes (top), but moves into the cytoplasm five days post-injury (middle, injured myocytes: eMHC, green). By 10 days post-injury (bottom), TDP-43 repairs back to the nucleus. [Courtesy of Vogler et al., Nature, 2018.]
Damage to neurons from TDP-43’s extranuclear wanderings was a predominant theme at this meeting. And yet, Roy Parker of the University of Colorado, Boulder, offered a counternarrative, at least in muscle cells. As recently published, Parker showed that TDP-43 not only forms amyloid-like structures with RNA in the cytoplasm, but suggested that these little nuggets serve a purpose during development and regeneration (Vogler et al., 2018).
Parker reasoned that RNA’s notorious tendency toward self-assembly made it surprising, if anything, that granules chock-full of entangled RNAs do not continually choke the cytoplasm. RNA-binding proteins clearly maintain order, Parker said, and nowhere is there more of a need for this than in muscle cells. Myocytes boast the longest-known RNA message in the transcriptome, a 100 kb monster encoding the giant muscle protein titin, and they have to deliver such RNAs over long distances to form super structures such as the sarcomere, a highly organized collection of bundled fibers. What’s more, TDP-43 is known to play a pivotal role in muscle development and maintenance, and cytoplasmic accumulation is a feature of degenerating muscles.
Parker found that when muscle fibers develop in mice, TDP-43 moves from the nucleus of myocytes into the cytoplasm, where it binds numerous mRNAs. From within the tube-like myocytes, TDP-43 formed rings around growing myofibers. Once the sarcomere had formed, TDP-43 returned to the myocyte nucleus. The same was true in adult myocytes spurred to regenerate after an injury: TDP-43 sprang out of the nucleus, RNA in tow, until the sarcomere was repaired.
Closer inspection of these transient, high-molecular-weight TDP-43-RNA structures brought up a resemblance to amyloid oligomers. Diffraction patterns indicated β-sheet content on par with oligomers made of α-synuclein and Aβ, yet none of the steric zipper motifs found in amyloid fibrils. The A11 antibody, known to bind a range of oligomers but not fibrils, latched onto these so-called “myogranules” as well (Apr 2003 news). Notably, the TDP-43/RNA structures contained some, but not all, components of stress granules. Myogranules were also distinct from the stress granules that formed when the researchers subjected myofibers to stress conditions. Parker identified numerous TDP-43 binding sites within known myocyte mRNAs, including titin, suggesting several TDP-43 molecules might dot the surface of mature mRNAs in the cytoplasm.
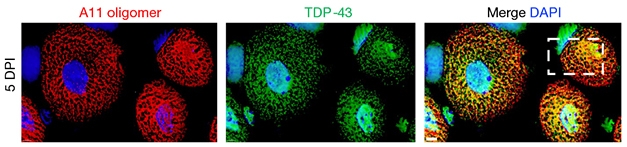
Oligomers with a Purpose. Five days post-injury, oligomers recognized by the A11 amyloid antibody (red) co-localize with TDP-43 (green) in the cytoplasm of myocytes. [Courtesy of Vogler et al., Nature, 2018.]
Parker proposed that these transient structures might keep RNA from getting entangled during times of intense gene-expression changes, e.g., during development or regeneration. He envisions TDP-43 amyloid-like structures as protective cages that shuttle RNA to distant sites for local translation. In myopathies, when muscles are constantly trying to repair themselves, an overabundance of these structures, coupled with a failure to clear them, could seed a pathological aggregation. Parker found numerous A11-positive TDP-43 aggregates in muscle samples from people with necrotizing myopathy, a disease in which myocytes are in a perpetual state of repair.
Could similar mechanisms be at play in neurons? Following neuronal injury, or in the face of chronic neurodegeneration, neurons might require the services of TDP-43 to shuttle numerous mRNAs out to axons, dendrites, and other distant locations for translation. Could such myogranules jump across neuromuscular junctions, and seed pathological aggregation in motoneurons? No evidence exists to suggest this could be the case, Parker told Alzforum. Even so, he suggested that an interesting future experiment would be to see if TDP-43 containing myogranules can also initiate a trans-synaptic propagation of TDP-43 aggregates.
At the meeting, Virginia Lee of the University of Pennsylvania in Philadelphia showed data that ranked TDP-43 itself—not myogranules—among the proteins that can propagate via proteopathic spread. Alzforum covered her group’s paper, which reported that aggregates extracted from postmortem brains of people with FTD, when injected into TDP-43 transgenic mouse brain, seeded trans-synaptic propagation (Oct 2018 news).
Wolozin, for one, was fascinated by Parker’s findings. They mesh with his and others’ observation that except in cultured cell lines, cytoplasmic TDP-43 generally does not coalesce into stress granules under physiological conditions. It appears more diffusely distributed. Other researchers were intrigued by Parker’s discovery of a normal, physiological purpose for cytoplasmic TDP-43 aggregates. Paul Taylor of St. Jude’s Children’s Hospital in Memphis, Tennessee, noted that this role as a protector of RNA molecules could also be true of other RNA-binding proteins, such as FUS, which have been spotted accompanying RNA to distant reaches of neurons. “When conditions change, such as when cells become stressed, then these protective structures could join up with stress granules or other membraneless organelles,” Taylor suggested. If this continues unabated, toxic aggregation might ensue, he added. For more on FUS, see Part 3 in this series.—Jessica Shugart
References
News Citations
- Going the Distance: FUS Travels to Terminals, Drops Off RNA
- Forming FUS Droplets with a Flash of Light
- Amyloid Oligomer Antibody—One Size Fits All?
- TDP-43 Joins Cell-To-Cell Propagation Gang
Paper Citations
- Vogler TO, Wheeler JR, Nguyen ED, Hughes MP, Britson KA, Lester E, Rao B, Betta ND, Whitney ON, Ewachiw TE, Gomes E, Shorter J, Lloyd TE, Eisenberg DS, Taylor JP, Johnson AM, Olwin BB, Parker R. TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature. 2018 Nov;563(7732):508-513. Epub 2018 Oct 31 PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.