Alpha-Synuclein Stymies Amyloid Plaques, but Worsens Synaptic Damage
Quick Links
Like a zealous chaperone at a middle school dance, α-synuclein may block Aβ from clumping together. A study led by Melanie Meyer-Luehmann at Ludwig-Maximilians University in Munich reported that α-synuclein prevented plaques from forming in mouse models harboring both α-synuclein and Aβ pathology. However, in the presence of α-synuclein, the reduction in plaque burden offered no relief from neuronal damage—in fact, the mixed mice suffered more synaptic deficits than animals harboring only a single pathology. Published June 22 in Nature Medicine, the findings could offer mechanistic insight into neurodegeneration in people burdened with mixed pathologies.
“The in vivo experiments demonstrate quite clearly that not only does α-synuclein not enhance Aβ deposition, but actually inhibits,” commented Kelvin Luk of the University of Pennsylvania in Philadelphia, who was not involved in the study. “This is somewhat counterintuitive and quite interesting given that Aβ and α-synuclein pathology frequently co-occur in human disease.”
Aβ and α-synuclein are the culprits implicated in AD and synucleinopathies such as Parkinson’s disease and dementia with Lewy bodies (DLB), respectively. While misbehaving forms of the two proteins are associated with different neurodegenerative diseases, their occurrence in the same person tend to be the rule rather than the exception: Postmortem, a majority of people with DLB also harbor AD pathology, and around half of AD patients have Lewy bodies (see McKeith et al., 2004; Hamilton et al., 2000).
Alpha-synuclein is predominantly an intracellular protein, while Aβ roams outside of cells, raising the question of how the two could interact. Long ago, one study reported spotting α-synuclein fragments mingling with Aβ plaques (Ueda et al., 1993). Other researchers have suggested that synthetic forms of the two proteins form a complex on the neuronal membrane that creates pores and messes with calcium intake (see Sep 2008 news). Other studies suggest that the two pathologies egg each other on, with α-synuclein promoting Aβ pathology and vice versa (see Oct 2001 news; Feb 2008 news; May 2010 news). Both α-synuclein and Aβ reportedly act as prions, as misfolded forms of each protein can corrupt healthy versions into misfolding, as well.
Meyer-Luehmann’s initial goal was to test whether α-synuclein could “cross-seed” Aβ and trigger its aggregation. First author Teresa Bachhuber and colleagues injected APPPS1 mice that had not yet developed plaques with brain extracts from A30P-α-syn mice, which express a pathogenic mutation of human α-synuclein and are riddled with Lewy bodies. The injection did not trigger Aβ plaque formation, although injecting APPPS1 mice with brain extracts from plaque-bearing APPPS1 mice did seed plaques. The cross-seeding failed to occur even when the researchers injected brainstem homogenate (which is loaded with Lewy bodies) from 21-month-old A30P-α-syn mice, or when the researchers injected brain homogenate from a patient with severe DLB. Preformed fibrils of recombinant human α-synuclein also failed to induce Aβ plaque formation.
The researchers were initially flummoxed by the lack of cross-seeding. Then they decided to monitor the interaction between Aβ and α-synuclein throughout the entire plaque formation process. They crossed the APPPS1 mice with the A30P-α-syn mice. Compared to APPPS1 mice, the double-transgenic animals had fewer hippocampal plaques at 4 months of age, according to staining with different Aβ antibodies as well as thiazin red, which labels dense core plaques. In keeping with their reduced plaque burden, double-transgenic mice had higher levels of CSF Aβ42 than APPPS1 mice. Despite their reduced plaque load, brain extracts showed the double-transgenic mice had just as much insoluble Aβ as the APP/PS1 mice. In young mice that had not yet deposited plaques, levels of soluble Aβ, as well as APP mRNA, were the same in double- and single-transgenic mice. Together, these results suggest that α-synuclein somehow interferes with the formation of Aβ plaques, but does not disrupt Aβ production or even the formation of insoluble species.
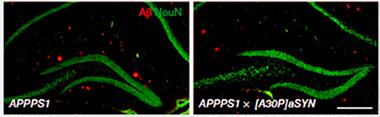
Synuclein Blocks Plaque.
APPPS1 mice (left) have more plaques (red) in the hippocampus than mice also expressing a pathogenic form of α-synuclein (right). [Courtesy of Nature.]
The researchers further defined the inhibitory role of α-synuclein in Aβ plaque formation by performing additional seeding experiments. They mixed together extracts from A30P-α-syn mice and APPPS1 mice, and injected them into APPPS1 mice that had not yet developed plaques. Few plaques formed in this situation. However, when the researchers first depleted α-synuclein from the injection mixture, the extracts triggered robust plaque formation. Notably, the researchers got a similar result when they injected APPPS1 mice with AD brain extracts (which triggered plaques), or brain extracts from a person with mixed AD/DLB pathology (which did not trigger plaques).
In another line of experiments, the researchers grafted embryonic stem cells from wild-type or A30P-α-synuclein mice into the brains of APP/PS1 mice. Although the transplanted cells did not express human APP, plaques from the surrounding host tissue ultimately infiltrated the wild-type graft. However, very few plaques crept into the A30P-α-syn grafts, indicating that α-synuclein warded off plaque formation.
To take a closer look at the interaction between α-synuclein and Aβ, the researchers mixed the proteins together and monitored Aβ aggregation by electron microscopy as well as ThT fluorescence. They found that whether α-synuclein harbored the pathogenic A30P mutationo or not, it inhibited the formation of fibrils that did occur when Aβ42 was agitated alone for 24 hours.
What were the functional consequences of this inhibition of plaque formation by α-synuclein? To address this question, the researchers compared the density of dendritic spines and the concentration of synaptophysin in APPPS1 versus double-transgenic mice. They found that before plaques appeared in either strain, the double-transgenic mice had lower levels of both, suggesting that their synapses were worse off than those in the APPPS1 mice. The researchers point out that these findings support observations in people with mixed DLB/AD pathology, who develop cognitive deficits earlier than their AD-only counterparts (see Olichney et al., 1998).
Meyer-Luehmann speculated that α-synuclein’s meddling in plaque formation could elevate other species of Aβ known to cause synaptic damage. “If α-synuclein interacts with pre-fibrillar forms of Aβ so plaques cannot form, then perhaps toxic forms of Aβ are accumulating,” she told Alzforum. She plans to conduct behavioral studies to test whether the synaptic problems translate into cognitive ones, and biochemical studies to confirm that α-synuclein blocks Aβ through a direct interaction.
Brit Mollenhauer of Paracelsus-Elena-Klinik in Kassel and University Medical Center Göttingen, both in Germany, commented that the results suggest that Lewy bodies and amyloid plaques may be the wrong pathologies to focus on in terms of neurodegeneration. Other forms of the proteins may truly cause the synaptic deficits that lead to cognitive decline. “The findings could also underline that we need to target not only one but several pathologies with our immunotherapies (and maybe not Aβ plaques at all) to save synapses and to have a clinical benefit in our trials,” she added.
“Taken together, their data suggest that various forms of these molecules may form co-deposits that prevent fibril formation while increasing neurotoxicity in the brain of transgenic mice,” commented Martin Ingelsson of Uppsala University in Sweden. He added that because injections in the study were done on both sides of the brain, it remains unclear whether the injected material spread to other regions.
“This is a careful and elegant study. It illustrates that not only can Aβ influence α-synuclein’s state of aggregation, but that synuclein can also influence solubility and fibrillation of Aβ,” wrote Eliezer Masliah of the University of California, San Diego. The findings further confirm that Aβ and α-synuclein share common pathogenic pathways relevant to AD, DLB and PDD, he wrote.
Ian McKeith of Newcastle University in England commented that when it comes to human disease, the relationship between plaques and Lewy bodies remains obscure. He cited his recent study that assessed the accuracy of diagnoses in autopsy-confirmed DLB patients. The researchers concluded that DLB symptoms correlated with higher amounts of Lewy body pathology and less severe neuritic plaque pathology (see Tiraboschi et al., 2015). However, it was difficult to conclude much about the relationship between the two pathologies.
Another study reported that people with mixed DLB/AD pathology and people with pure AD had similar amounts of plaques in the medial temporal lobe, which would be inconsistent with Meyer-Luehmann’s findings in animals, McKeith wrote (see Gentleman et al., 1992). However, Meyer-Luehmann cited yet another study that indicated that people with mixed pathology had fewer plaques than people with AD only (see Heyman et al., 1998). The jury may still be out on how the two proteins affect each other in people, but perhaps the animal studies will continue to shed some light on the murkiness of mixed pathology. —Jessica Shugart
References
News Citations
- Guilt by Association?—Aβ, α-Synuclein Make Mixed Oligomers
- Aβ Abets α-Synuclein
- Synuclein Surprise: Toxicity Linked to Aβ, Plaques?
- Triple Trouble: AD Mice Decline Faster With Lewy Bodies
Research Models Citations
Paper Citations
- McKeith I, Mintzer J, Aarsland D, Burn D, Chiu H, Cohen-Mansfield J, Dickson D, Dubois B, Duda JE, Feldman H, Gauthier S, Halliday G, Lawlor B, Lippa C, Lopez OL, Carlos Machado J, O'brien J, Playfer J, Reid W, . Dementia with Lewy bodies. Lancet Neurol. 2004 Jan;3(1):19-28. PubMed.
- Hamilton RL. Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000 Jul;10(3):378-84. PubMed.
- Uéda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993 Dec 1;90(23):11282-6. PubMed.
- Olichney JM, Galasko D, Salmon DP, Hofstetter CR, Hansen LA, Katzman R, Thal LJ. Cognitive decline is faster in Lewy body variant than in Alzheimer's disease. Neurology. 1998 Aug;51(2):351-7. PubMed.
- Tiraboschi P, Attems J, Thomas A, Brown A, Jaros E, Lett DJ, Ossola M, Perry RH, Ramsay L, Walker L, McKeith IG. Clinicians' ability to diagnose dementia with Lewy bodies is not affected by β-amyloid load. Neurology. 2015 Feb 3;84(5):496-9. Epub 2014 Dec 31 PubMed.
- Gentleman SM, Williams B, Royston MC, Jagoe R, Clinton J, Perry RH, Ince PG, Allsop D, Polak JM, Roberts GW. Quantification of beta A4 protein deposition in the medial temporal lobe: a comparison of Alzheimer's disease and senile dementia of the Lewy body type. Neurosci Lett. 1992 Aug 3;142(1):9-12. PubMed.
- Heyman A, Fillenbaum GG, Gearing M, Mirra SS, Welsh-Bohmer KA, Peterson B, Pieper C. Comparison of Lewy body variant of Alzheimer's disease with pure Alzheimer's disease: Consortium to Establish a Registry for Alzheimer's Disease, Part XIX. Neurology. 1999 Jun 10;52(9):1839-44. PubMed.
Further Reading
Papers
- Masliah E, Rockenstein E, Inglis C, Adame A, Bett C, Lucero M, Sigurdson CJ. Prion infection promotes extensive accumulation of α-synuclein in aged human α-synuclein transgenic mice. Prion. 2012 Apr 1;6(2):184-90. PubMed.
- Kallhoff V, Peethumnongsin E, Zheng H. Lack of alpha-synuclein increases amyloid plaque accumulation in a transgenic mouse model of Alzheimer's disease. Mol Neurodegener. 2007;2:6. PubMed.
Primary Papers
- Bachhuber T, Katzmarski N, McCarter JF, Loreth D, Tahirovic S, Kamp F, Abou-Ajram C, Nuscher B, Serrano-Pozo A, Müller A, Prinz M, Steiner H, Hyman BT, Haass C, Meyer-Luehmann M. Inhibition of amyloid-β plaque formation by α-synuclein. Nat Med. 2015 Jul;21(7):802-7. Epub 2015 Jun 22 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Pennsylvania
The in vivo experiments demonstrate quite clearly that not only does α-synuclein not enhance Aβ deposition, but actually inhibits it. This is somewhat counterintuitive and quite interesting given that Aβ and α-synuclein pathology frequently co-occur in human disease. It also raises the question of where such an interaction would occur, given that one is mainly an extracellular peptide while the other intracellular. The in vitro experiments point toward their interacting as monomers/oligomers and it will be interesting to see if the hybrid oligomers postulated by the authors are indeed a highly toxic species.
University of Toronto
This is an intriguing study. Already in 2007, Hui Zheng and colleagues (Kallhoff et al., 2007) showed that the lack of α-synuclein reduced plaque pathology and increased synaptophysin levels in Aβ transgenic mouse brains. Now, Bachhuber and colleagues have performed a series of intelligent in vitro and in vivo studies to further elucidate the interplay between Aβ and α-synuclein. Taken together, their data suggest that various forms of these molecules may form co-deposits that prevent fibril formation and increase neurotoxicity in the brains of transgenic mice. However, since the intracerebral injections of α-synuclein-containing samples were performed bilaterally, we don’t know if global seeding effects also occurred. Given the recent studies demonstrating contralateral propagation of α-synuclein, more far-reaching intermolecular seeding effects could indeed be possible.
References:
Kallhoff V, Peethumnongsin E, Zheng H. Lack of alpha-synuclein increases amyloid plaque accumulation in a transgenic mouse model of Alzheimer's disease. Mol Neurodegener. 2007;2:6. PubMed.
Newcastle University,
Little is known about the mutual trajectories of α-synuclein and Aβ accumulation in vivo in humans. Longitudinal imaging studies do not yet exist. We recently examined the clinical phenotype (cognitive fluctuation, recurrent visual hallucinations, parkinsonism, and REM sleep behavior disorder) in 64 autopsy-confirmed cases of dementia with Lewy bodies (DLB). Neocortical Aβ deposits were quantified using the BA4 antibody. Each brain was also staged for the degree of neuritic plaque pathology according to CERAD criteria using the Gallyas silver technique, and for the degree of neurofibrillary tangle pathology by the Braak method using the AT8 antibody. Lower frequencies of core clinical features of DLB, resulting in lower accuracy of its clinical diagnosis, were associated with decreasing Lewy body distribution (p < 0.0001) and with increasing neuritic plaque pathology (p = 0.035), but not with the number of Aβ plaque deposits (Tiraboschi et al., 2015). We concluded that the molecular relationship between LB- and AD-type pathology remains obscure. It may be that Aβ deposition is the primary event in both AD and DLB and that preferential downstream effects are either tau aggregation, resulting in a clinical phenotype more like that of AD, or α-synuclein aggregation, resulting in a clinical phenotype more like that of DLB. In some individuals, these overlapping pathologies result in an ambiguous clinical profile.
This report by Bachhuber and colleagues of synuclein inhibiting amyloid deposition is not immediately compatible with this model, nor with the fact that when amyloid is seen in DLB cases (about 50 percent of the time) using in vivo brain PET, levels are generally in the AD range. Why amyloid is deposited in some, but not all DLB cases remains unanswered.
References:
Tiraboschi P, Attems J, Thomas A, Brown A, Jaros E, Lett DJ, Ossola M, Perry RH, Ramsay L, Walker L, McKeith IG. Clinicians' ability to diagnose dementia with Lewy bodies is not affected by β-amyloid load. Neurology. 2015 Feb 3;84(5):496-9. Epub 2014 Dec 31 PubMed.
Make a Comment
To make a comment you must login or register.