ALS Mutations Stiffen Profilin, Steer Protein Toward Aggregation
Quick Links
Several different mutations in the tiny profilin-1 are responsible for some familial forms of amyotrophic lateral sclerosis. According to computational models published in the August 30 Scientific Reports, these variants restrict the protein’s flexibility, straining its relationship with actin and potentially many other partners. The mutations also make the protein more hydrophobic, coaxing it to aggregate. The authors, led by Mahmoud Kiaei at the University of Arkansas for Medical Sciences in Little Rock, propose that the structural constraints lead to a toxic mix of misfolding, aggregation, and loss of function that eventually fell motor neurons during aging.
- The G118V and T109M mutations weaken binding of profilin-1 to actin and poly-L-proline, respectively.
- Both mutations make profilin-1 more rigid and hydrophobic.
- The mutations likely promote aggregation.
Mutations in profilin-1 (PFN1) account for 1 to 2 percent of familial ALS cases (Jul 2012 news; Jun 2015 news). At just 140 amino acids in length, the protein contains only two interaction domains. With one, it binds actin monomers and plays an essential role in their polymerization and in the formation of the cytoskeleton. With the other, PFN1 latches onto proteins that contain poly-L-proline (PLP) motifs. These proteins carry out a variety of functions. Profilin binds upward of 50 partners, suggesting its influence stretches well beyond the cytoskeleton. Cell culture and mouse models indicate that ALS-causing mutations in the PFN1 gene destabilize the protein, promote its aggregation, and even instigate the aggregation of TDP-43, another player in ALS pathogenesis (Oct 2016 news; Fil et al., 2017; Tanaka et al., 2016). However, researchers have yet to nail down which of these mechanisms ultimately steers motor neurons toward their demise.
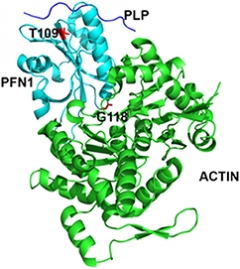
Small Socialite.
PFN1 (light blue) interacts with PLP (top, blue strand) and actin (green). The G118 and T109 residues mutated in ALS interact with actin and PLP, respectively. [Courtesy of Kiaei et al., Science Reports, 2018.]
Kiaei and colleagues investigated how two different mutations changed the protein’s shape. Using a previously published crystal structure of PFN1 in complex with actin and PLP, they simulated the structural and functional consequences of the glycine-118-valine (G118V) and threonine-109-methionine (T109M) mutations (Ferron et al., 2007). According to the crystal structure, G118 interacts directly with actin via a hydrogen bond, and the simulations indicated that switching this residue to valine, a larger nonpolar amino acid, would restrict PFN1’s flexibility and weaken its association with actin. Furthermore, the conformational changes caused by the valine substitution led to greater exposure of an adjacent valine residue—V119. Together, these exposed sticky residues boosted PFN1’s overall hydrophobicity, a property known to increase the likelihood of aggregation.
The T109 residue resides adjacent to, but is not part of, the PLP binding site. However, the computational simulations revealed that substituting the threonine with the bulkier methionine would shift the conformation of residues that do contact PLP, leading to weaker binding. Similar to the scenario with G118V, the T109M mutation also made PFN1 more rigid and hydrophobic. The findings suggest that a combination of rigidity and hydrophobicity would weaken the protein’s associations with its binding partners, and promote its aggregation.
How might these structural shifts lead to motor neuron demise that occurs only with aging? Kiaei explained that in the G118V mouse model he developed, aggregates of PFN1 start accumulating in motor neurons prior to the onset of motor symptoms. He proposed that early in life, neurons manage to recognize and dispose of the misfolded protein translated from the mutant copy of PFN1. During this time, the cells make do with their single good copy of the gene. However, at some point in the aging process, motor neurons fail to clear the accumulating misfolded protein, which then aggregates and ensnares other proteins, perhaps including the normal copy of PFN1. The mutant protein may also wreak havoc by virtue of weakened associations with its binding partners, he added, but he thinks toxic gain of function primarily drives ALS in these familial cases.
Kiaei noted that as part of the protein’s normal life cycle, PFN1 is tagged for disposal via ubiquitin. He speculated that the increased rigidity of the protein caused by the mutations may limit its efficient labeling and disposal.
In his lab and through a company he founded called Rock Gen Therapeutics, Kiaei is testing whether small molecules that stabilize PFN1 can prevent the toxic cascade. He is using a similar strategy to stabilize proteins that aggregate in other neurodegenerative diseases as well.—Jessica Shugart
References
News Citations
- Profilin Gene Is Actin’ in ALS
- Mutations Open Cavity in Profilin, Destabilize the ALS-Linked Protein
- Profilin 1 Mutant Mouse—a New Model for ALS?
Research Models Citations
Paper Citations
- Fil D, DeLoach A, Yadav S, Alkam D, MacNicol M, Singh A, Compadre CM, Goellner JJ, O'Brien CA, Fahmi T, Basnakian AG, Calingasan NY, Klessner JL, Beal FM, Peters OM, Metterville J, Brown RH Jr, Ling KK, Rigo F, Ozdinler PH, Kiaei M. Mutant Profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum Mol Genet. 2017 Feb 15;26(4):686-701. PubMed.
- Tanaka Y, Nonaka T, Suzuki G, Kametani F, Hasegawa M. Gain-of-function profilin 1 mutations linked to familial amyotrophic lateral sclerosis cause seed-dependent intracellular TDP-43 aggregation. Hum Mol Genet. 2016 Apr 1;25(7):1420-33. Epub 2016 Jan 28 PubMed.
- Ferron F, Rebowski G, Lee SH, Dominguez R. Structural basis for the recruitment of profilin-actin complexes during filament elongation by Ena/VASP. EMBO J. 2007 Oct 31;26(21):4597-606. Epub 2007 Oct 4 PubMed.
Further Reading
Papers
- Ingre C, Landers JE, Rizik N, Volk AE, Akimoto C, Birve A, Hübers A, Keagle PJ, Piotrowska K, Press R, Andersen PM, Ludolph AC, Weishaupt JH. A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol Aging. 2012 Nov 8; PubMed.
Primary Papers
- Kiaei M, Balasubramaniam M, Govind Kumar V, Shmookler Reis RJ, Moradi M, Varughese KI. ALS-causing mutations in profilin-1 alter its conformational dynamics: A computational approach to explain propensity for aggregation. Sci Rep. 2018 Aug 30;8(1):13102. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Tokyo Metropolitan Institute of Medical Science
The mechanism of how PFN1 mutations cause familial ALS is still unclear, but these mutations are autosomal-dominant and mutants are destabilized and prone to form aggregates (Boopathy et al., 2015). Therefore, gain of toxic function of mutant PFN1 would at least partially contribute to the onset or progress of the disease. In fact, we previously showed the toxicity of PFN1 mutants in disorganizing TDP-43, which is the central molecule of ALS (Tanaka et al., 2016; Matsukawa et al., 2016). Interestingly, recent preprint shows that PFN1 mutants (C71G, G118V, E117G) disrupts the liquid-liquid phase separation that organizes membrane-less organelles, while wild-type PFN1 doesn’t change such dynamics (Kang et al., 2018). The mechanism whereby ALS-linked PFN1 induces seed-dependent intracellular TDP-43 aggregation remains unknown, but misfolded PFN1 might be a key to induce conformational changes in TDP-43. This study by Kiaei et al., shows that the change of flexibility of the actin or PLP binding site might increase the propensity for PFN1 to aggregate. In other words, the possible changes on the binding site in PFN1 might cause PFN1 aggregation. Considering that TDP-43 accumulates in sporadic ALS patients, PFN1 abnormality might not be restricted to familial ALS.
References:
Boopathy S, Silvas TV, Tischbein M, Jansen S, Shandilya SM, Zitzewitz JA, Landers JE, Goode BL, Schiffer CA, Bosco DA. Structural basis for mutation-induced destabilization of profilin 1 in ALS. Proc Natl Acad Sci U S A. 2015 Jun 30;112(26):7984-9. Epub 2015 Jun 8 PubMed.
Tanaka Y, Nonaka T, Suzuki G, Kametani F, Hasegawa M. Gain-of-function profilin 1 mutations linked to familial amyotrophic lateral sclerosis cause seed-dependent intracellular TDP-43 aggregation. Hum Mol Genet. 2016 Apr 1;25(7):1420-33. Epub 2016 Jan 28 PubMed.
Matsukawa K, Hashimoto T, Matsumoto T, Ihara R, Chihara T, Miura M, Wakabayashi T, Iwatsubo T. Familial Amyotrophic Lateral Sclerosis-linked Mutations in Profilin 1 Exacerbate TDP-43-induced Degeneration in the Retina of Drosophila melanogaster through an Increase in the Cytoplasmic Localization of TDP-43. J Biol Chem. 2016 Nov 4;291(45):23464-23476. Epub 2016 Sep 15 PubMed.
Kang J, Lim L, Song J. Misfolded proteins share a common capacity in disrupting LLPS organizing membrane-less organelles. bioRχiv. 2018
Northwestern University, Feinberg School of Medicine
In this paper, Kiaei and colleagues have taken a unique approach to investigate how mutations in the profilin gene affect the three-dimensional structure of the protein and its interaction with binding partners. This study, even though mainly performed in silico, offers insight into potential interaction problems that occur due to mutations. Studies like this are important for drug discovery efforts, as they reveal potential sites for drug-target interaction. I wish we had similar studies for all other protein products of genes that are mutated in ALS patients. I congratulate Kiaei and colleagues for this insightful and out-of-the-box research endeavor.
University of Arkansas for Medical Sciences
I appreciate the interesting points in two separate comments by Drs. Tanaka and Ozdinler.
The published work of Tanaka’s group on the role of mutant PFN1 disorganizing TDP-43 is of great interest and is consistent with the evidence that we presented that in our mutant PFN1 mouse model that TDP-43 was abnormally distributed and appeared aggregated spinal cord (Fil et al., 2017). The suggestion by Tanaka that PFN1 abnormality could occur in sporadic ALS is actually very reasonable. This is a highly important hypothesis that can be tested if suitable PFN1 antibodies become available.
By pointing to the main outcome of this work and how it can be applied toward drug discovery efforts, the comment and feedback by Ozdinler on the nicely written report by Jessica Shugart emphasize the value and impact of a study like this and are highly encouraging. We are currently pushing forward with promising follow-on studies toward developing compounds to reverse the changes that we described in our paper and block the aggregation of mutant PFN1.
References:
Fil D, DeLoach A, Yadav S, Alkam D, MacNicol M, Singh A, Compadre CM, Goellner JJ, O'Brien CA, Fahmi T, Basnakian AG, Calingasan NY, Klessner JL, Beal FM, Peters OM, Metterville J, Brown RH Jr, Ling KK, Rigo F, Ozdinler PH, Kiaei M. Mutant Profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum Mol Genet. 2017 Feb 15;26(4):686-701. PubMed.
Make a Comment
To make a comment you must login or register.