Could Extra Granulins Be the Problem in Progranulin Insufficiency?
Quick Links
People with only one good copy of progranulin typically develop frontotemporal dementia (FTD), leading scientists to suspect that a half-dose of progranulin is insufficient for neural health. However, a paper in the June 24 Journal of Neuroscience explores another side of the story: the granulins that result when progranulin gets cleaved. In nematodes, these products elevate levels of TDP-43, escalating toxicity, according to researchers at the University of California in San Francisco. While loss of full-length progranulin might still be important in the pathogenesis of FTD, the results imply that the granulins, too, spark or stoke the disease, said senior author Aimee Kao. However, she has not yet worked out how or if less progranulin leads to extra granulin activity.
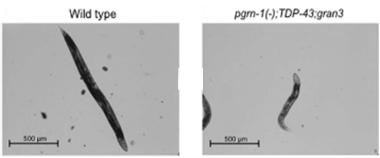
Wimpy Worms:
Nematodes toting granulin, but not progranulin, plus human TDP-43 are puny. [Courtesy, with permission, of Salazar et al., The Journal of Neuroscience, 2015.]
Progranulin participates in a variety of cellular processes, including inflammation, apoptosis, and stress responses. While people with one progranulin gene develop FTD, a pair of siblings homozygous for a progranulin loss-of-function mutation developed a different condition—neuronal ceroid lipufuscinosis (NCL). They both became ill in their 20s, with symptoms including seizures, vision loss, and ataxia (Smith et al., 2012). Researchers are still unsure how PGRN mutations cause either disease. Complicating their investigations, cellular proteases cleave progranulin into seven granulins—six with a characteristic sequence containing 12 cysteine residues, and one petite granulin with only half that motif. It has been difficult to pin down the role of granulins, Kao said, because there are so many and they are so closely entwined with progranulin. In the new study, first author Dominique Salazar used Caenorhabditis elegans to address two key questions. How do progranulin heterozygotes differ from null mutants? And how do granulins contribute to disease?
Since progranulin mutations lead to TDP-43 proteinopathy, Salazar started with a worm strain expressing human, wild-type TDP-43 in its 302 neurons. In a commonly used motility assay for worms, the TDP-43 strain squirmed only about three-quarters as often as wild type.
If loss of progranulin were the main problem in FTD, the authors would expect that crossing the TDP-43 worms with a progranulin null strain (with neither progranulin nor granulins) would worsen the movement disorder. However, it made no difference. Next, Salazar mimicked the human FTD condition, leaving the worms with only one copy of progranulin (and thus some granulins as well). This made the worms worse; they thrashed only about one-third as often as wild-type animals. Since the presence of granulins was one major difference between the PGRN hetero- and homozygotes, the authors turned their attention to those fragments.
Worm progranulin contains three granulins. To focus solely on those, Salazar created transgenics expressing individual granulins 1, 2, or 3, but no full progranulin, and crossed them with the TDP-43 strain. While she saw little effect from granulin 1, TDP-43 worms with granulins 2 or 3 were puny (see image above). They also thrashed less, again performing at about one-third of wild-type levels in the motility assay. “Granulins are synergistically toxic with TDP-43,” Kao concluded.
How? While the researchers have not worked out the full mechanism, they found a clue. Worms with granulin 2 or 3 possessed more TDP-43 protein, but not TDP-43 mRNA, than other strains. Kao speculated that the granulins might interfere with lysosomal degradation of TDP-43, leading to accumulation of the protein.
Next, the authors examined granulins in tissues from people who died of FTD with TDP-43 proteinopathy. They generated an antibody specific to one human granulin, granulin E (homologous to worm granulin 3), and detected more of that fragment in the diseased regions of those brains than in unaffected regions or control brains. The results hint that granulins contribute to the human disease, either alone or in concert with haploinsufficiency of full-length progranulin.
If that is so, it suggests that something tips the balance between progranulin and granulins, with too many granulins piling up when there is only one copy of the progranulin gene. One other study also suggested this balance might be important, since the granulins counteract progranulin’s effects in wound healing (Zhu et al., 2002). However, Kao’s results imply that having only one copy of progranulin leads to more granulins. That is a pretty big leap, said Erik Roberson of the University of Alabama in Birmingham, who was not involved in the study. It is not clear if or how this would occur in either worms or people, he said. Kao speculated that if an organism only has one copy of progranulin, but normal levels of the proteases that cleave it, that half-supply of progranulin might be over-processed into excess granulins. Researchers have shown that the blood and cerebrospinal fluid of people heterozygous for PGRN mutations contain less than half the normal amount of the progranulin (Finch et al., 2009; Ghidoni et al., 2008; Sleegers et al., 2009).
“The most interesting aspect is the difference between [progranulin] knockouts and heterozygotes in their ability to exacerbate TDP-43 toxicity,” commented Li Gan of the Gladstone Institutes in San Francisco, who was not involved in the work. One recent article supported that finding, reporting that zebrafish lacking progranulin have no disease phenotype (Solchenberger et al., 2015). Kao and colleagues observed the same in their worms that had no progranulin or TDP-43 transgene, she said. Together, these studies suggest that alone, loss of progranulin may not be toxic.
They also imply that having one copy of progranulin is quite different from having none at all, raising an important question about null models for disease. “Is that [null] really modeling FTD, or is it modeling NCL?” asked Roberson. “And what is the relationship between these two diseases?” This would not be the first mutant gene to cause different phenotypes in the homozygous and heterozygous states. In the case of TREM2, a double whammy causes FTD or Nasu-Hakola disease, while a single bad allele increases risk for Alzheimer’s and ALS (see Oct 2012 news; Nov 2012 news; Feb 2014 news).
Kao, too, raised questions about the use of null models to understand FTD. “We would suggest people be cautious about directly interpreting the null progranulin animals to the haploinsufficiency state,” she said. Jiou Wang of Johns Hopkins University in Baltimore thought that null models have value, but that researchers should consider heterozygous models as well. He added, “The involvement of the granulins are definitely worth looking into in other models and in people.” Wang was not involved in Kao’s study.
The results also have implications for FTD therapeutics in which researchers hope to boost progranulin levels (see Nov 2014 news). More progranulin, especially in an inflamed environment that promotes granulin production, might lead to more granulins and worsen the disease, Kao speculated.—Amber Dance
References
Alzpedia Citations
News Citations
- Mutations in TREM2 Cause Frontotemporal Dementia
- Enter the New Alzheimer’s Gene: TREM2 Variant Triples Risk
- TREM2 Variant Doubles the Risk of ALS
- Progranulin-Boosting Drug Moves into Phase 2 for Frontotemporal Dementia
Paper Citations
- Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, Sims KB, Lewis J, Lin WL, Dickson DW, Dahl HH, Bahlo M, Berkovic SF. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 2012 Jun 8;90(6):1102-7. PubMed.
- Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument-Bromage H, Tempst P, Wright CD, Ding A. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002 Dec 13;111(6):867-78. PubMed.
- Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R, Bisceglio G, Rovelet-Lecrux A, Boeve B, Petersen RC, Dickson DW, Younkin SG, Deramecourt V, Crook J, Graff-Radford NR, Rademakers R. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 2009 Mar;132(Pt 3):583-91. PubMed.
- Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology. 2008 Oct 14;71(16):1235-9. PubMed.
- Sleegers K, Brouwers N, Van Damme P, Engelborghs S, Gijselinck I, van der Zee J, Peeters K, Mattheijssens M, Cruts M, Vandenberghe R, De Deyn PP, Robberecht W, Van Broeckhoven C. Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol. 2009 May;65(5):603-9. PubMed.
- Solchenberger B, Russell C, Kremmer E, Haass C, Schmid B. Granulin knock out zebrafish lack frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis pathology. PLoS One. 2015;10(3):e0118956. Epub 2015 Mar 18 PubMed.
Further Reading
Papers
- Filiano AJ, Martens LH, Young AH, Warmus BA, Zhou P, Diaz-Ramirez G, Jiao J, Zhang Z, Huang EJ, Gao FB, Farese RV, Roberson ED. Dissociation of frontotemporal dementia-related deficits and neuroinflammation in progranulin haploinsufficient mice. J Neurosci. 2013 Mar 20;33(12):5352-61. PubMed.
- Premi E, Padovani A, Borroni B. Frontotemporal Lobar Degeneration. Adv Exp Med Biol. 2012;724:114-27. PubMed.
- Liscić RM. Frontotemporal dementias: update on recent developments in molecular genetics and neuropathology. Arh Hig Rada Toksikol. 2009 Mar;60(1):117-22. PubMed.
- Neumann M, Tolnay M, Mackenzie IR. The molecular basis of frontotemporal dementia. Expert Rev Mol Med. 2009;11:e23. PubMed.
- Mackenzie IR. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007 Jul;114(1):49-54. PubMed.
News
- Does Progranulin Play Both Sides in AD and FTD?
- FTD Risk Factor Confirmed, Alters Progranulin Pathways
- Could Sortilin Be a Sweet Spot for FTD Therapy?
- Back to Basics? Boosting pH Puts Cells in Progranulin-Pumping Mode
- Meet Progranulin, The Biomarker—A Simpler Story?
- Salzburg: New Proteins Redefine Frontotemporal Dementias
Primary Papers
- Salazar DA, Butler VJ, Argouarch AR, Hsu TY, Mason A, Nakamura A, McCurdy H, Cox D, Ng R, Pan G, Seeley WW, Miller BL, Kao AW. The Progranulin Cleavage Products, Granulins, Exacerbate TDP-43 Toxicity and Increase TDP-43 Levels. J Neurosci. 2015 Jun 24;35(25):9315-28. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Colorado
This is an interesting study that attempts to get at the underlying mechanism(s) by which mutations in the progranulin gene (PGRN) cause familial Frontotemporal Dementia (FTD). Progranulin encodes an evolutionarily conserved precursor protein that can be proteolytically processed to produce seven distinct granulin peptides. Frontotemporal dementia caused by PGRN mutations is characterized by cytoplasmic TDP-43 inclusions in affected neurons, a pathology that is also seen in sporadic cases of FTD and ALS. PGRN disease mutations are autosomal dominant yet are caused by a loss-of-function, unlike other familial FTD mutations (e.g. in Tau and TDP-43). This result suggests that just having less PGRN is sufficient to cause neurodegeneration. However, having less is different than having none, and extrapolating from complete knockout models could be misleading.
Salazar et al. specifically examined the role of C. elegans progranulin in the toxicity of a transgenic C. elegans model expressing human wild-type TDP-43. As observed by other research groups, expression of human TDP-43 in C. elegans neurons resulted in movement defects. These movement defects were not altered in worms homozygous for a deletion of their endogenous progranulin gene (pgrn-1). Intriguingly, motor defects were enhanced in worms heterozygous for the pgrn-1 deletion (i.e., equivalent to the situation in humans patients). This result suggests that the phenotypic interaction between TDP-43 and PGRN may actually require some progranulin expression. These researchers went on to show that expression of two individual granulin peptides (granulins 2 and 3) could exacerbate TDP-43 toxicity and enhance TDP-43 protein levels, while a third granulin (granulin #1) had no effect.
How can this curious result be explained? The authors suggest that the processing of progranulin might be finely regulated by the ratio of progranulin produced to the proteolytic enzymes that act on it. Theoretically, a reduction of progranulin "substrate" could lead to more or different proteolytic processing if there is no coordinated change in the processing enzymes. It is tempting to speculate that alterations in processing could alter the ratio of full progranulin to granulin or the ratio of individual granulins produced. (Note that in the case of brain-derived neurotrophic growth factor, proBDNF has effects opposite to processed BDNF.) If individual granulins have competing functions (e.g., might granulin 1 oppose the actions of granulins 2 and 3?), changing the ratios of the granulins could have physiological effects that could not be observed in the homozygous deletion strain.
The above hypothesis suggests numerous follow-up experiments. The suite of granulins actually produced in people homozygous or heterozygous for the PGRN gene has yet to be determined, although this research group has now developed an antibody specific for human granulin "E". The C. elegans model should allow combinatorial expression of individual granulins and the identification of regulators of progranulin processing through forward genetic screens. Finally, if the "altered proccessing" model is correct, one might expect genes encoding the relevant processing factors to be fingered in genome-wide association studies of FTD, or possibly directly implicated in familial FTD cases.
Make a Comment
To make a comment you must login or register.