Alzheimer’s Transmission Between People? Amyloid Plaques in Hormone Recipients Hint at Prion-like Spread
Quick Links
Consensus is building among researchers that misfolded Aβ peptides can transmit their bad behavior when transferred from human to mouse or animal to animal—but could this transmission happen between people? According to a small study published in Nature on September 9, the possibility is real. Drawing on autopsy tissue, researchers led by John Collinge and Sebastian Brandner at University College London reported the presence of abundant Aβ plaques in the brain’s parenchyma and blood vessels in a handful of people who received injections of human growth hormone extracted from cadavers (c-HGH) decades ago. The patients had all died between the ages of 36 and 51 of Creutzfeldt-Jakob Disease (CJD), which they contracted from the injections. None had developed symptoms of Alzheimer’s disease or cerebral amyloid angiopathy (CAA). Moreover, their brains lacked the telltale tau pathology that defines AD. Even so, the presence of Aβ pathology in such a young group of people set off alarm bells. Other researchers called for further investigation into the possibility that, like prions, perhaps Aβ seeds can indeed spread from one person to another under certain conditions.
“This is a pathogenic principle we discovered in animals, and this study suggests it could happen in humans,” said Mathias Jucker of the German Center for Neurodegenerative Diseases and Hertie Institute for Clinical Brain Research in Tübingen, who was not involved in the study. “The conclusion of transmission is not yet proven, but surely this finding warrants heightened concern and further study,” he said. Jucker published a paper on the same day in Nature Neuroscience. It revealed that so-called Aβ seeds remained able to induce amyloidosis even after lying dormant for six months in a mouse brain. These findings add to mounting evidence that such seeds are robust. They also allude to the extraordinarily long incubation period of Alzheimer’s disease. Studies indicate that amyloid plaque and tau tangle pathology start to manifest 25 or more years before the emergence of cognitive symptoms.
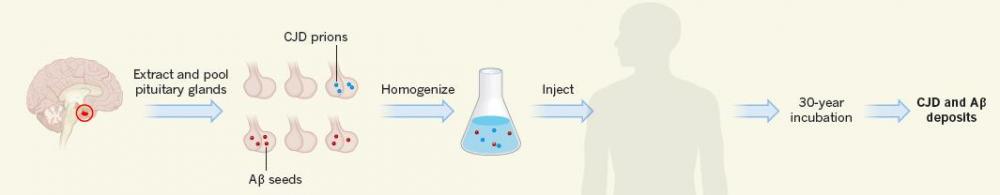
Stealth Seeds. Besides prions, an unknown fraction of human growth hormone extracts isolated from pituitary glands may also have contained Aβ seeds. Recipients of these treatments later developed both CJD and Aβ deposition. [Courtesy of Mathias Jucker and Lary Walker, News & Views, Nature 2015.]
As misfolded versions of normal proteins, prions corrupt properly folded proteins and propagate pathology. The prion protein PrP is the infectious agent in prion diseases in animals and humans, in whom it can cause CJD. Some people contracted a variant of this fatal neurodegenerative disease by eating contaminated meat, and fears of an impending epidemic rocked the United Kingdom and ravaged its beef industry at the turn of this century. CJD can also arise sporadically or due to mutations that render PrP prone to misfolding.
Transplants from donors with CJD or exposure to contaminated surgical equipment represent another route. Called iatrogenic CJD (iCJD), this version of the disease afflicted 226 people who had received intramuscular injections of c-HGH derived from pituitary glands of cadavers. Most of these patients had received the injections as children to help them grow taller. Scores of pituitary glands excised from the brains of organ donors were pooled and served as the source for the hormone, which was partially purified with the biochemical methods of the day. In the United Kingdom alone, an estimated 400,000 pituitary glands were used for this purpose. An estimated 30,000 patients received repeated injections of this preparation from 1959 until 1985, when cases of CJD surfaced and manufacturers switched to recombinant production (see Will, 2003; and Brown et al., 2012).
Athletes and body builders have long used HGH to boost performance. Despite the International Olympic Committee’s ban on doping with HGH, some athletes continued to purchase the cadaveric form of the hormone on the black market long after the more expensive, recombinant version had supplanted it for medical purposes (see Deyssig and Frisch, 1993; Sonksen, 2001; Holt and Sonksen, 2008). Pituitary glands illegally taken from cadavers may have provided a source for the hormone, as one Russian media report of a pituitary-gland-smuggling operation documented in 2000 (see Moscow Times, 2000).
Other neurodegenerative disease-associated proteins, including Aβ, tau, and α-synuclein, have also been shown to propagate and cause pathology in a mode that some researchers call “prion-like.” For example, Aβ seeds extracted from human AD brain or transgenic AD mouse brains and transferred to mice expressing human APP induced AD pathology in those recipient mice (see Jul 2009 news and Oct 2011 news). Given the similarities with prions, researchers have wondered whether Aβ or other neurodegenerative-disease proteins could also spread via c-HGH injections or other procedures. In 2013, researchers led by John Trojanowski at the University of Pennsylvania in Philadelphia concluded that such transmission was unlikely. Sifting through death certificates from nearly 800 people who had died since receiving the c-HGH injections from the U.S. National Hormone and Pituitary Program (NHPP) about 30 years earlier, they found no evidence of increased rates of death due to AD or PD in this cohort (see Feb 2013 news).
For the current study, first author Zane Jaunmuktane and colleagues examined whether c-HGH recipients who later contracted CJD also harbored AD pathology. By 2012, 65 out of 1,800 people in the United Kingdom who had been previously treated with c-HGH had developed CJD. In the United Kingdom, a majority of patients with prion disease have been recruited into the National Prion Monitoring Cohort (NPMC) study, including 22 c-HGH recipients who developed iCJD in recent years. The researchers conducted in-depth autopsies on eight of these patients, who had developed iCJD an average of 25 years after c-HGH treatment.
In addition to the expected CJD pathology, Aβ deposits riddled the brain parenchyma of four of the patients. An additional two patients had smaller, focal Aβ deposits, and one had a small amount; only one of the eight patients’ brains was devoid of Aβ pathology. Three of the four patients with substantial amyloidosis also had cerebral amyloid angiopathy (CAA), a condition marked by damage to brain vessels that can trigger bleeding or strokes. Genetic testing indicated that none of the eight people harbored mutations in any of 16 genes associated with early onset AD, CAA, or other neurodegenerative diseases, and none carried the ApoE4 allele.
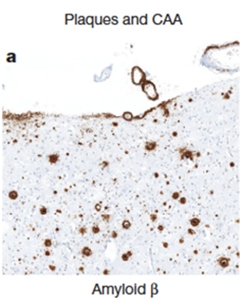
Seeds Sown?
Diffuse amyloid plaques speckle the brain of a c-HGH recipient. Cerebral amyloid angiopathy is apparent around blood vessels (top).
“These are highly unusual findings, as you wouldn’t expect this level of Aβ deposition at this age,” Collinge told reporters at a press briefing. For example, one neuropathology study of people without CJD reported that just 10 out of 290 people aged 36 to 51 had comparable Aβ pathology (see Braak and Braak, 1997). Still, the small sample size precludes definitive statements about whether the frequency of Aβ pathology in these eight patients was truly abnormal, commented Trojanowski.
To determine whether pituitary glands could have been the source of Aβ contamination, the researchers examined autopsies from 49 people with cerebral Aβ pathology, and found AD pathology in the pituitary glands of seven. This indicated that pituitary glands were a potential source of Aβ seeds. Both autopsy and amyloid PET studies have shown that the prevalence of brain AD pathology rises with age (e.g., Price et al., 2009; Villemagne et al, 2013). Many pituitary donors were elderly, suggesting that, unbeknownst to investigators at the time, the prevalence of amyloid pathology among them may have been significant.
One alternative potential explanation for the Aβ pathology in the present study is that CJD pathology somehow could have triggered amyloid deposition. A connection between CJD and Aβ has been reported in some studies but not others (see Tousseyn et al., 2015; Hainfellner et al., 1998). To investigate this, the researchers looked to autopsy data from more than 100 people in the NPMC who suffered from forms of CJD not transmitted though c-HGH, such as sporadic or variant CJD and inherited prion diseases. They found no significant Aβ pathology in the 19 patients who had been between 36 and 51 at death. Two of the 35 patients between the ages of 52 and 60 harbored significant Aβ pathology, but they carried the ApoE4 allele, which is known to bring on Aβ at an earlier age. This suggested to the authors that CJD, at least in its other forms, does not hasten Aβ deposition.
Furthermore, the researchers found no spatial overlap between CJD pathology and Aβ pathology in the brains of their iCJD cohort. While these findings do not prove that the patients acquired Aβ pathology directly from the c-HGH stocks rather than as a consequence of CJD, they support that conclusion, Collinge said.
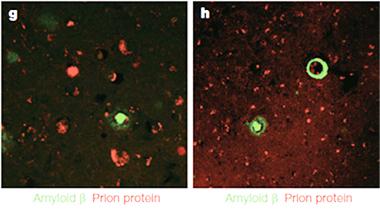
No Overlap.
Aβ (green), in the form of plaques (left) or CAA (right), was separate from prion plaques (red) in the brains of c-HGH recipients.
Marc Diamond of the University of Texas Southwestern Medical Center in Dallas agreed that transmission of Aβ from contaminated c-HGH extracts was the likeliest explanation for the results. “The implication, of course, is that many other proteins besides PrP could potentially be infectious,” Diamond wrote. Nevertheless, he downplayed potential risks. “I still highly doubt that in standard medical practice it would be possible to transmit pathology efficiently between individuals. Since we are no longer doing these pooled tissue treatments, which in retrospect seem particularly foolhardy, I would not imagine that we will be seeing a high number of “infected” cases of AD, CAA, or PD,” Diamond wrote to Alzforum.
Importantly, tau pathology was absent from all eight of the iCJD patients autopsied by Collinge and colleagues. The researchers speculated that perhaps the patients would have developed tau pathology in later years, had they not succumbed to CJD in midlife. CSF biomarker studies suggest that Aβ changes precede tau, and human mutant APP transgenic mice develop CSF tau changes subsequent to CSF Aβ changes.
In a joint comment to Alzforum, Dominic Walsh and Dennis Selkoe of Brigham and Women’s Hospital in Boston cautioned against equating the presence of Aβ deposition with future AD. “This paper does not report the transmission of AD or AD pathology, rather it documents the detection of Aβ deposits in individuals at an age when normally such deposits are extremely rare,” they wrote. “The fact that the brains of these individuals did not contain neurofibrillary tangles indicates that these individuals did not have AD, and even if they had lived longer, it is uncertain that they would have developed AD.”
While the U.K. researchers cannot know whether the patients would have developed AD, they concluded that the severity of CAA in these patients alone was cause for concern, as they would have been at elevated risk for cerebral hemorrhages had they lived longer. Walsh and Selkoe agreed that potential fallout from CAA was more troubling than Aβ deposits.
What of the thousands of c-HGH recipients who did not develop iCJD? According to Collinge, approximately 1,500 people who received the injections in the United Kingdom are still alive. They were not formally contacted by the respective authority, the U.K. Department of Health, about the results of this study prior to its publication. “Many of these individuals unfortunately are going to find out about this from the media,” Collinge said in the press briefing. He added he would like to monitor these people for the emergence of AD pathology via PET scans or CSF biomarkers. Collinge invited concerned patients to contact his center; however, no plan for a formal study is yet in place. Approximately 1,800 patients in France and 7,700 patients in the United States also received c-HGH injections.
The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) retains records of recipients in the United States, but officials at NIDDK were unavailable for comment. Information for patients regarding risks associated with c-HGH injections can be found on the agency’s website.
Collinge told reporters that his lab is seeking permission to access archived batches of the c-HGH extracts that were injected into patients. He would like to test these batches for the presence of infectious Aβ seeds, however, he noted that the properties of such seeds are poorly understood and no sufficiently sensitive biochemical assay exists to date. The tried-and-true way to test an Aβ-containing mixture for infectivity is to inject animals and wait, sometimes as long as a year, for Aβ pathology to emerge.
Researchers are busy developing in vitro assays to obtain quicker results. Claudio Soto of the University of Texas in Houston has developed research-grade assays to detect Aβ oligomers in the CSF of AD patients, as have several other labs (see Mar 2014 news), but results are not broadly reproduced and it is not clear how those species relate to the Aβ seeds that propagate amyloid pathology. Similar to assays scientists developed to detect prions, Soto’s so-called protein-misfolding cyclic amplification (PMCA) assay works by allowing misfolded proteins to aggregate, then breaking them up into particles that seed further aggregates until detection becomes possible. Soto told Alzforum that his lab is working on expanding the assay to other sample types, including blood, and that he would test old c-HGH lots for the seeds if given access. Collinge said the development of such in vitro assays are also an active area of research in his lab.
A seed assay could be used to test whether Aβ seeds can reside outside the brain. If this is true, it would raise the question of whether recipients of transplant organs or blood products from donors unwittingly harboring AD pathology could potentially contract Aβ seeds.
What about Aβ seeds that stick to surgical equipment or to electrodes placed inside the brain, for example in the course of deep-brain-stimulation surgery or invasive EEG monitoring? Prion proteins reportedly have been transmitted in this way (see Belay et al., 2013; Thomas et al., 2013). Collinge told reporters he had previously developed a combination of enzymes and detergents that could destroy prions, but that the product was never manufactured for use in surgical settings, much to his disappointment. He said a similar product would likely destroy Aβ seeds as well.
According to previous work from Jucker’s lab, Aβ seeds are hardy. They retain their ability to trigger Aβ pathology in mice after a two-year soak in formaldehyde, and vanishingly small amounts of infectious particle can trip off the cascade (see Sep 2014 news). Like prions, Aβ seeds have a fondness for metal surfaces and are resistant to boiling (see Eisele et al., 2009; Meyer-Luehmann et al., 2006).
In his Nature Neuroscience paper, Jucker added further evidence of the potency of Aβ seeds. First author Lan Ye and colleagues injected 2.5 microliters of Aβ seed-containing tissue extract into two different mouse strains: APP23 mice that express human APP harboring an AD-associated mutation, and mice expressing no APP. One month later, the APP23 mice had higher levels of human Aβ than they had previously, indicating an early seeding response. The seeds stagnated in the mouse strain devoid of APP, as they had no template to corrupt and could not propagate. After 30 days, the seeds were undetectable by standard ELISA methods and immunoblotting, however, when the researchers employed the highly sensitive, bead-based platform Simoa, they were able to detect residual amounts of seed even after six months of dormancy in the APP-null mice. Despite their minuscule concentration, these seeds came alive again once researchers injected them into APP23 mice. Even after languishing in APP-null mice for half a year, the seeds ultimately triggered AD pathology upon secondary transmission into APP23 recipients.
Jucker’s latest finding indicates that Aβ seeds could be even more tenacious than prions, which reportedly lose their infectivity in mice sooner. Soto was surprised by the seeds’ persistence; however, he noted that newer, more sensitive detection methods may show prions to be more persistent than previously thought, as well.
What does all this say about precautions that should be taken during surgical and/or transplant procedures? All researchers and commentators interviewed for this story emphasized that much more research is needed to determine where Aβ seeds reside, whether they are truly capable of transmitting AD, and under what circumstances. The incubation period of up to 30 years makes these questions particularly difficult to answer. The present study could prompt researchers to systematically investigate the possibility of AD transmission, and to develop safety tools, such as assays for Aβ seeds and methods to destroy them. “Right now we don’t know if or how AD can be transmitted,” Soto said, “but it is better to be safe than sorry.”—Jessica Shugart
References
News Citations
- Aβ the Bad Apple? Seeding and Propagating Amyloidosis
- Seeds of Destruction—Prion-like Transmission of Sporadic AD?
- In Case You Wondered: Neurodegenerative Diseases Are Not Contagious
- Test Uses 'Seeding' to Detect Aβ Oligomers in Cerebrospinal Fluid
- Bad Seeds—Potent Aβ Peptides Instigate Plaques, Won’t Be Fixed
Research Models Citations
Paper Citations
- Will RG. Acquired prion disease: iatrogenic CJD, variant CJD, kuru. Br Med Bull. 2003;66:255-65. PubMed.
- Brown P, Brandel JP, Sato T, Nakamura Y, MacKenzie J, Will RG, Ladogana A, Pocchiari M, Leschek EW, Schonberger LB. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis. 2012 Jun;18(6):901-7. PubMed.
- Deyssig R, Frisch H. Self-administration of cadaveric growth hormone in power athletes. Lancet. 1993 Mar 20;341(8847):768-9. PubMed.
- Sonksen PH. Insulin, growth hormone and sport. J Endocrinol. 2001 Jul;170(1):13-25. PubMed.
- Holt RI, Sönksen PH. Growth hormone, IGF-I and insulin and their abuse in sport. Br J Pharmacol. 2008 Jun;154(3):542-56. Epub 2008 Mar 31 PubMed.
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997 Jul-Aug;18(4):351-7. PubMed.
- Price JL, McKeel DW, Buckles VD, Roe CM, Xiong C, Grundman M, Hansen LA, Petersen RC, Parisi JE, Dickson DW, Smith CD, Davis DG, Schmitt FA, Markesbery WR, Kaye J, Kurlan R, Hulette C, Kurland BF, Higdon R, Kukull W, Morris JC. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009 Jul;30(7):1026-36. PubMed.
- Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, Szoeke C, Macaulay SL, Martins R, Maruff P, Ames D, Rowe CC, Masters CL, Australian Imaging Biomarkers and Lifestyle (AIBL) Research Group. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013 Apr;12(4):357-67. Epub 2013 Mar 8 PubMed.
- Tousseyn T, Bajsarowicz K, Sánchez H, Gheyara A, Oehler A, Geschwind M, DeArmond B, DeArmond SJ. Prion Disease Induces Alzheimer Disease-Like Neuropathologic Changes. J Neuropathol Exp Neurol. 2015 Sep;74(9):873-88. PubMed.
- Hainfellner JA, Wanschitz J, Jellinger K, Liberski PP, Gullotta F, Budka H. Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol. 1998 Aug;96(2):116-22. PubMed.
- Belay ED, Blase J, Sehulster LM, Maddox RA, Schonberger LB. Management of neurosurgical instruments and patients exposed to Creutzfeldt-Jakob disease. Infect Control Hosp Epidemiol. 2013 Dec;34(12):1272-80. Epub 2013 Oct 24 PubMed.
- Thomas JG, Chenoweth CE, Sullivan SE. Iatrogenic Creutzfeldt-Jakob disease via surgical instruments. J Clin Neurosci. 2013 Sep;20(9):1207-12. Epub 2013 Jul 27 PubMed.
- Eisele YS, Bolmont T, Heikenwalder M, Langer F, Jacobson LH, Yan ZX, Roth K, Aguzzi A, Staufenbiel M, Walker LC, Jucker M. Induction of cerebral beta-amyloidosis: intracerebral versus systemic Abeta inoculation. Proc Natl Acad Sci U S A. 2009 Aug 4;106(31):12926-31. PubMed.
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006 Sep 22;313(5794):1781-4. PubMed.
External Citations
Further Reading
Papers
- Jucker M, Walker LC. Neurodegeneration: Amyloid-β pathology induced in humans. Nature. 2015 Sep 10;525(7568):193-4. PubMed.
- Goedert M. NEURODEGENERATION. Alzheimer's and Parkinson's diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science. 2015 Aug 7;349(6248):1255555. PubMed.
- Walker LC, Jucker M. Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci. 2015 Jul 8;38:87-103. Epub 2015 Mar 30 PubMed.
Primary Papers
- Jaunmuktane Z, Mead S, Ellis M, Wadsworth JD, Nicoll AJ, Kenny J, Launchbury F, Linehan J, Richard-Loendt A, Walker AS, Rudge P, Collinge J, Brandner S. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature. 2015 Sep 10;525(7568):247-50. PubMed.
- Ye L, Fritschi SK, Schelle J, Obermüller U, Degenhardt K, Kaeser SA, Eisele YS, Walker LC, Baumann F, Staufenbiel M, Jucker M. Persistence of Aβ seeds in APP null mouse brain. Nat Neurosci. 2015 Nov;18(11):1559-61. Epub 2015 Sep 9 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Brigham & Women's Hospital
Co-Director, Brigham and Women's Hospital's Ann Romney Center for Neurologic Diseases
This is an interesting and provocative study of brain and pituitary tissue from individuals who died of prion diseases. Between 1958 and 1985 a significant number of young individuals of short stature received growth hormone (GH) isolated from the pituitary glands of human cadavers. Sadly, a small number of these individuals subsequently developed Creutzfeldt-Jakob disease (CJD). The current study focuses on eight of these iatrogenic CJD (iCJD) cases, all of whom died between the ages of 36 and 51, and quantifies Aβ deposition in the brain parenchyma and vasculature. Three out of the eight had significant amounts of vascular deposits (known as congophilic or cerebral amyloid angiopathy, CAA). Moreover, four of the eight had significant Aβ deposits in the parenchyma. Nineteen individuals who died at comparable ages to the eight GH iCJD cases from prion diseases not associated with GH had little or no evidence of CAA or parenchymal Aβ. None of the eight GH iCJD cases (including the four with substantial Aβ) had mutations in genes believed to predispose to AD. Importantly, no neurofibrillary tangles, the other diagnostic lesion of AD, were detected in any of the four iCJD cases with abundant Aβ deposits.
Overall, the authors interpreted their data to demonstrate iatrogenic transmission of Aβ “pathology” in addition to iCJD. They encouraged: 1) watching for the appearance at autopsy of deposits of Aβ in currently still-living recipients of pituitary extracts; and 2) investigating whether other routes of tissue entry from one human into another, e.g., by transfused blood products or contaminated surgical instruments, could be associated with the development of Aβ deposits.
Some specific comments:
1. This paper does not report the transmission of AD or AD pathology, rather it documents the detection of Aβ deposits in individuals at an age when normally such deposits are extremely rare. The fact that the brains of these individuals did not contain neurofibrillary tangles (the other key histological marker of AD) indicates that they did not have AD. Even if they had lived longer, it is uncertain that they would have developed AD. Importantly, there was no mention about any AD-type peri-plaque neuritic dystrophy or microgliosis.
2. In terms of clinical relevance, the observed CAA may be of more concern than the parenchymal Aβ deposits. This is because the physical buildup of CAA can sometimes lead to micro-hemorrhages and ultimately stroke, whereas it is less clear that just Aβ deposits in the parenchyma have significant functional consequences. However, given that the chances of developing iCJD from prion-contaminated GH injections is low (3.6 percent for U.K. cases, and less than 1 percent for U.S. cases), and only three of eight iCJD cases examined here had CAA, it would appear that the likelihood of developing clinically noticeable CAA may be lower than developing clinical iCJD.
3. It remains unclear how much Aβ could actually have been expected to be present in the cadaveric pituitaries, how many of the donors would have been likely to have Aβ deposits in their pituitaries, whether the Aβ would be enriched in the final pooled extracts, and how many of the multiple human pituitary extracts each patient received would have contained Aβ. As the authors rightly point out, it will be important to examine the original GH extracts for the presence and forms of Aβ therein.
4. The results presented are consistent with a prior report of Alzheimer-type neuropathology in a 28-year old patient with iatrogenic Creutzfeldt-Jakob disease after grafting of cadaverous dura mater (Preusser et al., 2006).
We conclude that Jaunmuktane et al. have made an intriguing observation that definitely needs detailed follow-up epidemiologically. However, for the broad public, this report is by no means evidence for transmission of AD or even a definite risk of developing AD. This unusual, now-discontinued iatrogenic route via pituitary extracts means the risk to the general public of acquiring AD in any similar fashion is extremely low. In accord, the overwhelming majority of typical AD subjects have not had any exposure to tissue extract injections, dural grafts, or even blood product transfusions, indicating that this prion-like route of acquiring AD is a rare phenomenon, but nonetheless deserving of further scientific study.
References:
Preusser M, Ströbel T, Gelpi E, Eiler M, Broessner G, Schmutzhard E, Budka H. Alzheimer-type neuropathology in a 28 year old patient with iatrogenic Creutzfeldt-Jakob disease after dural grafting. J Neurol Neurosurg Psychiatry. 2006 Mar;77(3):413-6. PubMed.
University of Texas, Southwestern Medical Center
This is a simple but fascinating paper. It suggests to me that patients who received cadaveric growth hormone, and who came to autopsy because they were unlucky enough to have been exposed to PrP prions, may have received Aβ seeds along the way as well.
It is of course unclear whether PrP pathology could somehow trigger Aβ pathology. There is no good evidence for this. Thus, given how common both AD and CAA are, instead it seems likely that some of these donors’ brains (i.e., people who didn’t have prion disease, but did have Aβ accumulation) found their way into the pooled extracts that were given to the patients who ultimately came down with prion disease. This is supported by the investigators’ findings that pituitary glands feature Aβ accumulation.
The implication, of course, is that many other proteins besides PrP could potentially be infectious. I still highly doubt that in standard medical practice it would be possible to transmit pathology efficiently between individuals. Since we are no longer doing these pooled tissue treatments, which in retrospect seem particularly foolhardy, I would not imagine that we will be seeing a high number of “infected” cases of AD, CAA, or PD. But you never know.
University of Pennsylvania
The authors made clear they were referring to Aβ transmission and not transmission of Alzheimer’s disease, as none of the patients had clinical AD or the tangles that in addition to Aβ deposits are required to make a postmortem diagnosis of AD. Thus, it is important to emphasize that we reported in a study of nearly 7,700 similarly treated individuals in a NIH/CDC database that there were no cases of AD or PD among them (Irwin et al., 2013).
Briefly, we found no evidence to support concerns that AD proteins are transmitted from one person to another.
Irwin et al. analyzed data from an existing cohort of patients who had received cadaveric human growth hormone (c-hGH) extracted from postmortem pituitary glands via a national program, which was a beneficial treatment for stunted growth, before synthetic hGH was available. Nearly 7,700 patients were treated with c-hGH in the United States between 1963 and 1985. In the mid-1980s, more than 200 patients worldwide who had received c-hGH inadvertently contaminated with prion proteins from affected donor pituitary tissue went on to develop an iatrogenic form of Creutzfeldt-Jakob disease (i-CJD).
Since then, the cohort has been followed by NIH/CDC investigators to track any additional cases of CJD, with extensive medical histories for patients over the 30-plus years since the c-hGH therapy was stopped. In Irwin et al., we looked for signs of elevated risk of AD and PD among this group and found that none of the c-hGH recipients developed AD or PD, despite the presence of pathological AD (tau, Aβ) and PD (α-synuclein) proteins in pituitary glands from deceased subjects. This clarified that c-hGH recipients were most likely exposed to these neurodegenerative disease proteins linked to AD and PD, but this did not result in transmission of AD or PD from person to person in nearly 7,700 examined subjects in the database described above.
References:
Irwin DJ, Abrams JY, Schonberger LB, Leschek EW, Mills JL, Lee VM, Trojanowski JQ. Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol. 2013 Apr;70(4):462-8. PubMed.
Make a Comment
To make a comment you must login or register.