Was this year’s HAI conference a bit of a misnomer? You could say so, if only to tease. The ninth incarnation of this rapidly growing conference, held January 14 to 16 in Miami Beach, Florida, featured as much excitement about tau as about amyloid imaging, for which several PET tracers are FDA-approved. At HAI, a brand-new tau tracer called THK-5351 debuted and three tracers by Roche were poking out of the preclinical pipeline. The leader of the pack, the Phase 2 tracer T807/AV1451, dominated the agenda as data were pouring in on its performance in Alzheimer’s and non-AD tauopathies—most of it good, some still rough around the edges. Meeting abstracts are freely downloadable here.
Human Amyloid Imaging Meeting Was Abuzz With Talk of Tau
From January 14 to 16, the ninth annual Human Amyloid Imaging (HAI) conference unfolded in Miami Beach, Florida, with a record attendance of 340 scientists—and arguably a curious little identity problem. HAI began in 2007, soon after the discovery of Pittsburgh compound B (PiB), and quickly became the premier gathering for unpublished data sharing and frank, extensive discussion of the leading edge of all things amyloid PET. Truth be told, the ninth HAI meeting still was that. But the hottest ticket in Miami this time was tau. Talks and poster sessions, hallway and lunch conversations were most animated when it came to the rapid emergence of PET tracers for the live imaging of the second defining molecular pathology described by Alois Alzheimer in 1906.
HAI is co-organized by Keith Johnson of Massachusetts General Hospital, Bill Klunk and Chet Mathis at the University of Pittsburgh, and Bill Jagust at University of California, Berkeley.
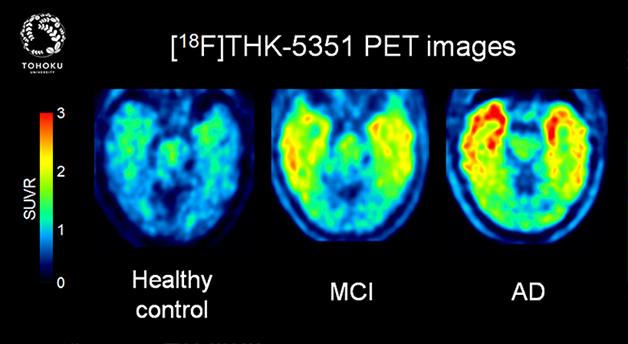
This new tau PET tracer shows low uptake in controls, intermediate uptake in mild cognitive impairment, and intense tau pathology spreading across the frontal and temporal cortex in Alzheimer’s disease. [Image courtesy of Nobuyuki Okamura, Tohoku University.]
With brain imaging for both amyloid plaques and tau tangles now at hand, scientists believe they will soon crack key questions in the field. Which sequence of events in the brain leads up to Alzheimer’s disease and other tauophathies? Which molecular pathology in which brain region causes which type of symptom, and how is normal brain aging different from neurodegenerative disease?
In AD in particular, scientists were excited about the notion that they will finally be able to understand how amyloid and tau pathology, as described for decades from postmortem brains, relate to each other during a person’s lifetime. The first data from people who have undergone both amyloid and tau scans strengthens the old concept that almost everyone develops circumscribed tau pathology with age. It impairs cognition slightly but falls well short of Alzheimer’s dementia. In the subset of people who also develop amyloid pathology, somehow this previously limited tau pathology intensifies, breaks out of the confines of the hitherto small brain regions it inhabited, and spreads catastrophically across the cortex. In other words, the idea is that amyloid pathology unleashes tau pathology, which then marches in lockstep with neurodegeneration and cognitive decline.
Clifford Jack of the Mayo Clinic in Rochester, Minnesota, summed it up this way: “Neuropathologists, for example Delacourte, Duyckaerts, and Price and Morris, proposed that medial temporal lobe age-related tauopathy happens in everybody, amyloid pathology happens in some, and amyloid accelerates tauopathy. PET imaging now validates what pathologists have said since the 1990s.”
Having stated the big idea, HAI attendees hastened to add that it’s extremely early days for tau PET. Altogether, fewer than 500 people have been scanned. For any given non-AD tauopathy, each research group has scanned barely more than a handful. And not all data fits. Off-target binding and some technical problems are cropping up that require more groundwork before tau PET will perform robustly and reproducibly in multicenter trials. “We are far from having a PiB for tau,” said Victor Villemagne of the University of Melbourne, Australia, referring to the amyloid tracer that is considered the gold standard on the Aβ side of things.
The tau PET field is expanding rapidly. The early lead belongs to 18F T807, a tracer Avid/Lilly bought from Siemens and renamed 18F AV1451. It is being studied by scientists at Avid and in academia, and scientists from six centers presented at HAI this year (see Part 2, Part 3, Part 4 , and Part 5 of this series). Having generated most of the early tau PET data known thus far, 18F AV1451 is already being prepared for use in several clinical trials, including the A4 Study. A second tracer called PBB3 is being studied by its developers at the National Institute of Radiological Sciences in Chiba, Japan, and by academic collaborators.
At HAI, a new tracer burst on the scene to general acclaim, and additional tracers are already nipping at its heels. Called 18F THK-5351, the new tracer is the latest in a series of PET compounds that researchers led by Nobuyuki Okamura at Tohoku University in Sendai, Japan, have been presenting over the past four years. Unlike its predecessors THK-523, THK-5105, and THK-5117, the new compound appears to have all the technical features nuclear medicine specialists are looking for, and some scientists at HAI declared 18F THK-5351 as being possibly the best of the bunch to date.
At HAI, Tohoku University’s Ryuichi Harada presented results from the first human trial of THK-5351. Harada acknowledged research support by GE Healthcare, which has previously collaborated with a Tohoku University biotech company on the university’s series of tau PET tracers. Scientists at HAI have therefore widely assumed that GE Healthcare will develop THK-5351 into a commercial tracer. GE Healthcare developed the FDA-approved amyloid tracer 18F flutemetamol, which grew out of 11C PiB.
Harada said that while previous Tohoku tracers visualized tau deposits in the brains of AD patients, their non-specific white-matter binding crowded out detection of small amounts of tau and made it impossible for the scans to be read visually. In contrast, when tested with autoradiography of AD brain sections, THK-5351 bound the same regions as tau antibodies, such as the hippocampus and inferior temporal cortex, with a high signal-to-noise ratio in gray versus white matter.
The scientists evaluated THK-5351 in 16 healthy controls, five people with mild cognitive impairment, and 13 AD patients. Two of them were scanned with both 5351 and the older 5117 tracer. THK 5351 entered the brain quickly, shooting up to peak retention five minutes after injection and leaving the brain by 90 minutes, Harada reported. Fifty minutes into this time window, the values for the SUVR—the most commonly used semi-quantitative measure of PET tracer uptake—stabilized at around 1.5 in the control group and 2.5 in the AD group. This is the best time window for imaging, Harada said.
The signal for THK-5351 has a larger dynamic range than that of its predecessors, Harada said. Together with the tracer’s quick clearance from white matter, this means the scans can be interpreted with the naked eye, an important criterion for future clinical use. THK-5351 distinguished AD from controls, and in MCI cases the amount of tau in signature regions such as the inferior temporal or parietal cortex was in-between. A voxel-by-voxel comparison of where PiB and THK-5351 bind across the Alzheimer’s brain aligned THK-5351 with the place where tau pathology would be expected according to Braak staging. THK-5351 does not bind Aβ, Harada said.
Adding his group’s data to the emerging sense that circumscribed tau pathology is quite common with age, Harada showed cross-sectional data suggesting that among the 16 cognitively healthy controls, THK-5351 uptake nudged upward with age in the hippocampus and, to a lesser degree, in the inferior temporal cortex.
On non-AD tauopathies, Harada showed first images of a patient with progressive supranuclear palsy, a rare disease that is currently a bit of a head-scratcher for PET imagers(see Part 4 of this series). The scan showed uptake in the patient’s midbrain, where it would be expected in this movement disorder. It also bound to those midbrain areas in micro-autoradiography of brain slices from a separate, postmortem PSP case.
“There are not a lot of imaging data on the new THK compound, but it appears that THK-5351 may be the best tau compound in AD we have seen in vivo thus far. It has great kinetics, low white-matter binding, and a large specific signal,” said Mathis, who discovered PiB (with Klunk) and is working on tracers for both tau and α-synuclein. Other PET experts agreed.
Pushing closely behind THK-5351 are candidates from the pharmaceutical and diagnostics giant Hoffmann-La Roche. Roche decided to develop its own tau PET tracer rather than trying to obtain AV1451 or another tracer for use in clinical trials of its investigational drugs targeted to either Aβ or tau. Besides tracking progression and drug response in therapeutic trials, companies are hoping the PET scans will become diagnostic tests in clinical practice.
Roche’s Michael Honer, Edilio Borroni, and colleagues collaborated with PET specialists led by Dean Wong at Johns Hopkins University School of Medicine in Baltimore. At HAI, they presented their in vitro and in vivo characterizations of three tracers called RO6931643, RO6924963, and RO6958948. If one of those early in vivo candidates shakes out as a keeper, it will get a more memorable name.
The Roche compounds displace the binding of the investigational Siemens tau tracer T808 to aggregated tau in human brain slices at Braak stages V/VI. Using both macro- and micro-autoradiography on human AD tissue, the Roche scientists showed that the binding of their candidate tracers co-localized with the immunohistochemistry of aggregated tau, but not Aβ. Then they shipped the precursor material to Baltimore, where Wong’s group radiolabeled and injected them into baboons. The baboons have no tau pathology, but are useful for measuring how quickly the tracers cross into the brain and wash out or become metabolized.
For their compounds, the Roche/Hopkins scientists reported high-affinity specific tau binding down to 5.5 nanomolars, strong selectivity over Aβ in human brain tissue, as well as low white-matter binding and rapid brain entry and washout in the baboons.
With that, Roche in August 2014 started a Phase 1 trial of all three tracer candidates in healthy controls and Alzheimer’s patients at Johns Hopkins. Importantly, Honer said, this trial will involve arterial blood sampling so that the tracer’s behavior over time can be understood in more depth than has been done with some previous tracers. Most years at HAI, debate flares up between scientists oriented toward the physics aspects of PET and researchers oriented more toward its medical applications. The former tend to argue the virtues of conducting dynamic imaging and detailed kinetic modeling studies of new tracers, while the latter emphasize getting the tracers out into the clinic and tend to favor simpler methods of static imaging. This technical debate surfaces when there is a new tracer, or later on when a tracer does not behave quite as scientists expected.
In essence, the choice boils down to using semi-quantitative measures such as standardized uptake values (SUV), which require only static scans, or quantitative measures such as distribution volumes, which require dynamic scans and blood samples, or binding potentials, which require dynamic scans but potentially not blood samples. The attraction of SUVs and static scans is that they make the data easier to acquire and analyze, but the researcher has to make more assumptions about how much non-specific binding or radiolabelled metabolites might contribute to the signal. Quantitative measures require fewer assumptions and therefore can serve to validate the tracer’s in vivo signal before simpler measures are used in the clinic, but they require acquisition and analysis that may not be available outside of research centers.
The debate in the amyloid and now also tau PET fields is about whether using the SUV is sufficient, or whether a tracer’s properties ought first to be validated with quantitative imaging, before picking methods that are feasible in routine practice. For PiB this has been done (e.g. Price et al., 2005; McNamee et al., 2009), and for the Roche tracers it is being done now; however, not all tracer developers believe this kind of work is necessary. For more on this issue, see Part 5 of this series.
For more information and freely downloadable program abstracts, see the conference website.—Gabrielle Strobel
Tau Tracer T807/AV1451 Tracks Neurodegenerative Progression
At the 9th Human Amyloid Imaging Conference, held January 14 to 16 in Miami Beach, Florida, 19 presentations by scientists at six different centers showcased data gathered thus far with T807. Now called AV1451, it is the most widely studied tau PET tracer to date, though others are pressing in fast (see Part 1 of this series). Overall, researchers showed largely converging results indicating that tau correlates more strongly than amyloid with neurodegeneration and cognitive decline in Alzheimer’s disease. Together, the data strengthen a growing sense that while amyloid and tau pathology first start up independently in separate regions in an aging person’s brain, the presence of amyloid somehow intensifies and accelerates an otherwise limited tauopathy. Exactly how that happens is one of the great mysteries in AD research these days, said Reisa Sperling of Harvard Medical School.
Researchers are starting to relate tau PET to CSF tau, and are beginning to track people over time with serial tau scans (see Part 3 of this series). Importantly, they are characterizing exactly what the tracer is binding. This is being done in postmortem brain slices from people who have not had a tau PET scan during life, because the tracers are so new that few cases have come to autopsy, the gold standard for this kind of validation. Scientists were pleased to find that T807/AV1451 does not appear to bind TDP-43 or a-synuclein, and hence will be helpful with the difficult task of differential diagnosis in the spectrum of frontotemporal and Lewy body dementias.
T807/AV1451 showed promise in detecting both atypical variants of AD and non-AD tauopathies; however, this story is only beginning to emerge. Scientists at HAI were puzzled by why T807/AV1451 appears to bind some regions considered devoid of tau pathology in AD (see Part 4 of this series). More worrisome to some, the tracer’s uptake in the cortex does not stabilize into a steady state but keeps climbing during the scanning period. This is not a problem for an up-down verdict of whether there is significant tau pathology in a given brain area, but it does raise questions about whether the measurement techniques for T807/AV1451 are quite ready for use in longitudinal and therapeutic studies that aim to quantify small changes over time (see Part 5).
“The pendulum has swung to one side, and we are rushing to use this tracer now in trials. I do not want it to swing back, but we should clean up those questions fast so the data we gather in trials are robust,” said Bill Klunk of the University of Pittsburgh Medical School.
Eli Lilly and Company owns the imaging company Avid Radiopharmaceuticals and bought the tau tracer T807 from Siemens (see Apr 2013 news). By now Avid has 11 clinical studies in various stages of planning or completion (see clinicaltrials.gov). At HAI, Avid’s Michael Pontecorvo presented the first cut of cross-sectional data, taken in September 2014, of an ongoing multicenter Phase 2 trial. This study has enrolled 230 cognitively normal and clinically affected volunteers and will follow some of them with repeat tau scans at nine and 18 months.
This first cut captured data on 156 people: 14 controls under 50 years old, 39 controls over 50, 70 people with mild cognitive impairment, and 33 with Alzheimer’s dementia. As a group, people with AD had more tau pathology than people with MCI, who had more than controls. At HAI, Pontecorvo’s colleague Mark Mintun told the audience that this largest study to date with AV1451 supports a simple graph by Pete Nelson and other leading neuropathologists in the field that was frequently cited at the HAI conference. It plots MMSE values at the time a person died against the amount of neurofibrillary tangles in their cortex to show that tau pathology correlates with cognitive impairment (Nelson et al., 2012). Tau PET now appears to bear this out.
Most importantly, perhaps, this ongoing AV1451 study points toward an interaction between amyloid and tau pathology. Like ADNI and several treatment trials, both the AD and MCI groups in this PET study contained some people who, despite their clinical diagnosis, had a negative florbetapir scan. These peoples’ cortical AV1451 uptakes were low, indicating that their symptoms were due to something other than Alzheimer’s. Among the cognitively normal volunteers, the picture was similar. In low-amyloid volunteers, T807/AV1451 uptake in the hippocampus did appear with age but stayed within a few small spots in medial temporal regions; in contrast, high-amyloid cognitively normal controls showed more tau pathology in a broader set of areas, particularly the inferior temporal cortex.
This hints that when there is amyloid, tau starts to aggregate beyond its initial confines in the medial temporal lobe. Other groups see this, as well. At HAI, Samuel Lockhart and others working with Bill Jagust at the University of California, Berkeley, showed in a series of 17 cognitively normal people that those without amyloid tended to accumulate tau pathology in the medial temporal lobe as they got older, but people with amyloid had tau pathology also spreading to the neocortex. Aaron Schultz and others in Keith Johnson’s group at Massachusetts General Hospital showed that among 75 older participants in the Harvard Aging Brain Study, the more amyloid a person had, the more tau pathology they also had in the inferior temporal cortex, an early “breakout” region of medial temporal lobe tau pathology.
The early confines of tau pathology in the medial temporal lobe are the hippocampus and parahippocampus, as well as entorhinal cortex. For cognitively normal people, tau spreading far beyond these small regions is bad news—and bad on a different scale than having a head full of amyloid, said Sperling. In previous work, she and colleagues had reported a cross-sectional link between the presence of brain amyloid and worse memory; however, her lab calls this the “weeny amyloid effect” because it is so small. Pathologists have claimed for years that neocortical tau pathology is more closely linked to cognitive impairment than amyloid, but the situation is complicated because some tau pathology also occurs with aging. This phenomenon recently got a new name, PART (Crary 2014). Now with both amyloid and tau tracers in hand, researchers can finally tease out in living people what matters most to cognition.
Sperling’s group reported on 97 older participants of the Harvard Aging Brain Study who had an AV1451/T807 scan within six months of cognitive testing. The scientists expressed cognition with a memory and an executive function score, each of which subsumed performance on three difficult tests. More tau accumulation in an early area of tau spread, the inferior temporal lobe, was associated with worse memory. The tau correlation was much stronger than that between amyloid burden and memory, and it held when adjusted for age, sex, and education. The same was not true of executive function in this study, Sperling said. “People with more amyloid also tend to have more tau, but the link between memory and tau is tighter in people with amyloid,” she said. Playing with these factors in a multiple regression model suggested, in essence, that tau mediates the association between amyloid and memory.
This would jibe with a new network analysis study that assigns different consequences to amyloid and tau pathology in several different lines of transgenic mice. At least in those models, amyloid touches off neuroinflammation, while tangles destroy synapses, according to this work by Frances Edwards and colleagues at University College London (see Jan 2015 news).
A definitive grasp of how amyloid, tau, and other factors drive AD requires longitudinal observation of where the pathologies accumulate locally, when they spread, and how that relates to neurodegeneration and evolving symptoms, Sperling said.—Gabrielle Strobel
Tau PET Fits With CSF, Grows Over Time, Picks up Frontotemporal Cases
How does tau PET fit in with more-established biomarkers of Alzheimer’s disease, such as the concentration of tau and phosopho-tau in the cerebrospinal fluid? For amyloid PET, the correlation with CSF Aβ was made early on (Fagan et al., 2006). It was subsequently confirmed to the point where amyloid PET and CSF Aβ are now seen as broadly equivalent for certain uses, for example, to screen prospective participants in a trial of an anti-amyloid drug. For tau PET, the first such data was presented at the 9th Human Amyloid Imaging conference, held January 14 to 16 in Miami Beach, Florida.
It came from the work of Jasmeer Chhatwal and colleagues at Harvard Medical School. Working with Keith Johnson and others, Chhatwal compared T807/AV1451 with CSF data in 31 older cognitively normal participants in the Harvard Aging Brain Study who had had a lumbar puncture and a tau PET scan within two years of each other. Comparing the CSF tau concentration to the T807/AV1451 signal in a host of different brain regions, the scientists spotted a statistically significant correlation in six regions. Intriguingly, these regions fall along the Braak staging route by which tau pathology is broadly suspected of spreading from its early site in the medial temporal lobe (MTL). They are the entorhinal and parahippocampal regions, and the inferior temporal, middle temporal, and superior temporal cortices.
Chhatwal and colleagues also related the tau PET signal to the participant’s CSF Aβ42 level. To do this with some spatial resolution, they drew up a matrix correlating the T807/AV1451 signal in each of eight regions along Braak’s proposed path of tangle spread to CSF tau, CSF phospho-tau, and CSF Aβ42. In this table a signal became apparent whereby the relationship between T807/AV1451 PET and CSF tau got weaker as it moved outward from the MTL and toward more distant regions of neocortex. In contrast, tau’s relationship to CSF Aβ42 persisted. This may be because the cognitively normal volunteers in this first sample had little tau pathology in those distant regions, but they did have amyloid there as part of the more widespread cortical deposition that is typical for amyloid pathology, Johnson said.
“If you believe the central tenet of the Braak staging system, that tau pathology spreads from the MTL to the neocortex, then you can interpret this data to mean that people with neocortical tau spread include individuals with high levels of brain Aβ deposition,” said Johnson, who presented the data on Chhatwal’s behalf. “As you move out along this path, you see a growing relationship with CSF Aβ and diminishing relationship with tau. “
This talk generated intense discussion. Clifford Jack of the Mayo clinic in Rochester, Minnesota, called it a “beautiful story.” Bill Jagust of the University of California, Berkeley, expressed hope that if these relationships show up even in cognitively normal people who have a relatively low tau signal, then scientists can hope to make robust and meaningful measurements of this sort in clinically affected people whose tau PET signal tends to be much higher.
In a separate talk, Hitoshi Shimada of the National Institute of Radiological Sciences in Chiba, Japan, presented binding data on his group’s tracer, PBB3. In this study, the PET signal did not correlate with CSF tau. This discrepancy could be due to differences in sample size, measurement techniques, or tracer characteristics, researchers said.
Does Tau PET Change Over Time?
As was the case with amyloid imaging, longitudinal observation will yield more powerful answers. With tau PET, repeat scanning is only just beginning, and at HAI, two groups presented their first case results. Mark Mintun of Avid Radiopharmaceuticals in Philadelphia is collaborating with Johnson and others to attempt to measure how the tau T807/AV1451 signal might evolve. Does it change slowly, as amyloid appears to do, making natural history and drug response tricky to quantify? Or does it grow fast, offering a wider range for researchers developing therapies to work with? To answer this question, the scientists first tried to get a sense of how reproducible T807/AV1451 measurements themselves are. In an initial sample of 21 older control, MCI, or AD cases, the difference between two scans taken within a month’s time was around 4 to 5 percent, Mintun told the audience. The intraclass correlation coefficient (ICC), used to express test-retest consistency, was above 0.93, Mintun said.
Mintun then showed longitudinal data from seven volunteers with MCI and one with AD. They had had two T807/AV1451 scans an average 14 months apart, along with two MMSE tests and a florbetapir amyloid scan. This is an exploratory study, Mintun noted. Two people had negative T807/AV1451 scans both times. One aced the MMSE both times, one did not; however, both were amyloid-negative. The remaining six people had a high amyloid burden. Their T807/AV1451 uptake grew between the first and second scans. By and large, their tau signal intensified and the distribution pattern expanded slightly. Five of the six performed lower on the MMSE the second time.
Mintun estimated a whopping 10 percent increase in uptake per year, though he emphasized that this preliminary measure will change as more data come in. “On the whole, it lines up. The tau SUVR went up as the MMSE went down in those folks who were amyloid-positive,” he told the audience.
Presenting the first data of his longitudinal T807/AV1451 work in non-AD dementias, Brad Dickerson of Massachusetts General Hospital showed two scans taken one year apart in a patient with a disease called nonfluent variant primary progressive aphasia (PPA). Dickerson not only saw the tau signal increasing but, tantalizingly, it looked as if tau pathology might be spreading through the language networks which functional connectivity MRI has implicated in this disease. For more news on tau PET in Alzheimer’s variants and frontotemporal dementia, see Part 4 of this series.—Gabrielle Strobel
What If It’s Not Garden-Variety AD? Telling Variants Apart by Where Tau Is
Scientists are increasingly confident that tau PET by and large matches up with expected regions and symptoms in Alzheimer’s disease, but does this hold true for diseases beyond typical AD, as well? This is an even younger area of tau PET exploration, because these cases are rarer, hence fewer patients have been scanned thus far. That said, at the 9th Human Amyloid Imaging conference last month in Miami Beach, Florida, several scientists showed early data hinting that tau PET might be useful here, too. Researchers were particularly excited by the prospect that tau PET might align quite well with other markers of neurodegeneration, particularly FDG PET. If larger studies bear out this trend, they may draw a sharp line between a specific protein pathophysiology and a person’s symptoms.
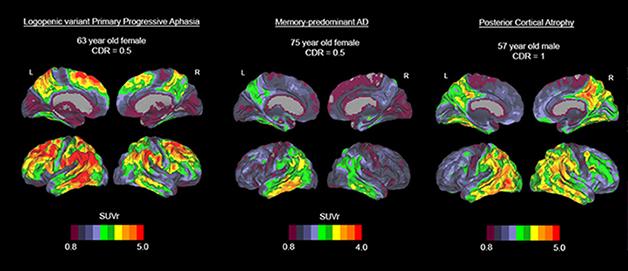
Three cases with distinct variants of Alzheimer’s disease show AV1451 uptake in distinct brain regions. A woman with PPA (left) shows an intense signal in language areas while her frontal and occipital areas are spared; a woman with mild typical AD (middle) appears to have tau pathology in the inferior temporal cortex; a man with posterior cortical atrophy (right) shows the strong signal in occipital and tempoparietal regions. [Image courtesy of Rik Ossenkoppele and Gil Rabinovici.]
At HAI, Eli Lilly’s Adam Schwarz told the audience that T807/AF1451 might be able to pick up two known variants of Alzheimer’s disease. One involves a hippocampal-sparing pattern, marked by fewer neurofibrillary tangles in the hippocampus and amygdala than typical AD, the other a so-called limbic-predominant variant marked by more tangles in the same areas. Concurring with this, Gil Rabinovici of the University of California, San Francisco, who won the 2015 Christopher Clark Award at this year's meeting, noted findings in his group that the regional pattern of tau, but not amyloid, PET tends to jibe with the regions thought to underlie the particular clinical symptoms in patients with atypical forms of AD.
As an example of Rabinovici’s group’s findings, Rik Ossenkoppele showed a poster about five patients with an AD variant called posterior cortical atrophy. People with PCA have visual and certain cognitive symptoms, but their memory initially stays intact. They have atrophy in the back areas of their brain, and Alzheimer’s pathology (see Jul 2012 conference news). Ossenkoppele and colleagues showed tau pathology in the expected occipitotemporal and –parietal regions in the back of the brain. Strikingly, in the PCA cases, and a handful of other AD-variant cases, the researchers saw virtual mirror images in the uptake of T807/AV1451 and 18F FDG, which measures neural activity. In other words, brain metabolism was abnormally low where tau pathology was high. This tight regional relationship was absent with amyloid PET, which is distributed more broadly across the association neocortex. Ossenkoppele published one PCA case report this month (Ossenkoppele et al., 2015).
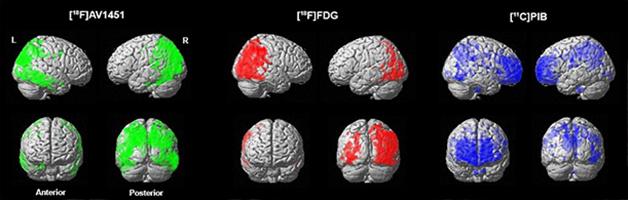
Voxel-wise comparisons between 16 controls and four patients with posterior cortical atrophy, a visual variant of Alzheimer’s disease, using 18F AV1451 PET (tau pathology, green), 18F FDG PET (glucose metabolism, red), and 11C PIB (amyloid deposition, blue). This figure demonstrates that tau aggregations strikingly mirror the posterior hypometabolic pattern on FDG-PET, while amyloid deposition is present in both clinically affected posterior regions and less-affected frontal areas. [Image courtesy of Rik Ossenkoppele and Gil Rabinovici.]
Tau PET carries much hope of enabling a molecular diagnosis in frontotemporal dementia. With this complex set of diseases, it is difficult for a clinician to predict which particular protein pathophysiology likely underlies a given patient’s clinical syndrome. Doing that, however, is important not only to managing the patient’s disease, but also for evaluating protein-specific therapies in clinical trials. Some FTD variants, for example progressive nuclear palsy (PSP), almost always have tau pathology. On the other hand, with behavioral variant FTD, the most common form, it’s about a 50-50 tossup between tau and TDP-43, and yet other FTDs fall in between. “Tau PET could really be a game-changer here,” Rabinovici said.
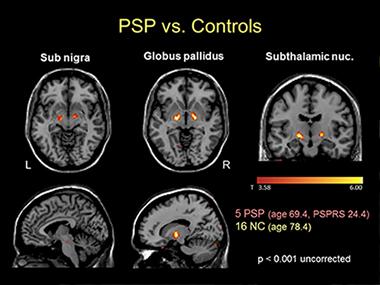
Voxelwise comparison of AV1451 retention in five people with progressive supranuclear palsy versus normal controls shows increased signal in globus pallidus, substantia nigra and subthalamic nucleus in this tau disease. [Image courtesy of Gil Rabinovici and Bill Jagust.]
At HAI, Rabinovici showed how T807/AV1451 performed for the first 13 non-AD cases scanned at UCSF. For example, the tau scan of a PSP case with a full deck of symptoms largely corresponded to the brain regions one would expect based on the published neuropathological staging system for the disease (Williams et al., 2007). However, the tracer also bound some of the same regions in cognitively normal controls. In a group of five PSP cases and 16 controls, the suspected PSP regions—substantia nigra, globus pallidus, and subthalamic nucleus—did have more binding in patients, but individual cases overlapped with controls. This could limit T807/AV1451’s ability to pick up early stages of PSP, Rabinovici said.
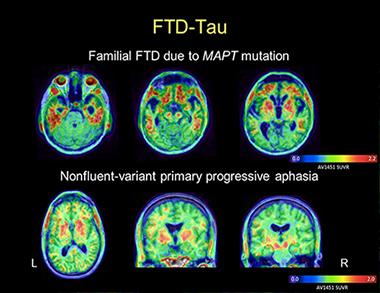
AV1451 images in a patient with behavioral-variant FTD due to MAPT mutation reveal tracer uptake in the frontal and anterior temporal lobes (top row). Images in a patient with nonfluent PPA show more asymmetric uptake in the left than the right peri-Sylvian cortex, a part of the brain’s language networks (bottom row). In both cases, the patterns of tracer uptake conform to the expected distribution of tau pathology as described in postmortem studies. [Image courtesy of Gil Rabinovici and Bill Jagust]
Of Rabinovici’s two primary progressive aphasia patients who have been scanned, one with the nonfluent variant (which is usually a tau disease) showed a plausible AV1451 distribution based on postmortem pathology of this syndrome. However, a person with semantic variant PPA had significant uptake, too, even though this disease is usually tau negative at postmortem. Likewise, for two bvFTD cases, one person’s tau scan instantly made sense and one did not. A case with a pathogenic tau mutation had a “spectacular” scan with intensive uptake in only the predicted regions, said Rabinovici. The other case was puzzling because the person carried a C9ORF72 repeat expansion and therefore would have been expected to have no T807/AV1451 uptake. Yet there it was—in degenerating white-matter tracts.
Similarly, of two suspected cerebral traumatic encephalopathy (CTE) cases, one showed T807/AV1451 uptake in regions matching neuropathology reported previously by Ann McKee of Boston University, and the other did not.
Brad Dickerson, who runs an FTD clinic at Massachusetts General Hospital, is finding much the same thing in that some scans fit the picture, some do not. At HAI, Dickerson, working with Stephen Gomperts and others, showed expected T807/AV1451 uptake in the basal ganglia, brainstem, and other predicted regions in PSP. Dickerson saw expected, asymmetric uptake in the dorsolateral prefrontal and insular cortex in nonfluent variant PPA, but also unexpected uptake in anterior temporal cortex in semantic variant PPA. Five tau mutation carriers, some asymptomatic and some with symptoms of bvFTD, retained T807/AV1451 in their frontal, insular, and anterior temporal cortices. In all cases, uptake and atrophy occurred in the same regions, Dickerson said.
For neuropathology correlations and technical research on the tracer’s behavior, see Part 5 of this series.—Gabrielle Strobel
Mixed Bag on Tau Tracer Validation, Kinetics: Some Things Fit, Some Don’t
Usually in PET development, before a new tracer is explored in many people and applied in clinical trials, it undergoes extensive validation to determine precisely where it is binding, and kinetic modeling studies to determine how it behaves in the human body over time. At least that is the standard sequence. For tau PET, the urgency of the disease burden—and the competitive advantage of having the first widely used tracer—is perceived to be so great that the leading tracer, T807/AV1451, is already being advanced into Phase 2 studies and readied for use in therapeutic trials before that groundwork is completed. Both fronts are moving in parallel. At the 9th Human Amyloid Imaging conference, held January 14 to 16, scientists reported results of this ongoing work, and debated them extensively.
Several groups presented largely converging results of binding studies in human postmortem brain tissue. Marta Marquie, who works with Teresa Gomez-Isla at Massachusetts General Hospital in Boston, used three different methods to characterize T807/AV1451 in brain slices of people who had died with Alzheimer’s; a tauopathy such as Pick’s disease, progressive supranuclear palsy (PSP), or corticobasal degeneration (CBD); an α-synucleinopathy such as dementia with Lewy bodies (DLB) or multiple system atrophy; or with TDP-43 inclusion pathology. First, Marquie used a fluorescent analogue of T807 and the antibody PHF1 directed against phosphorylated tau. Both bound to neurofibrillary tangles and dystrophic neurites in AD. Both also bound to a variety of expected structures in non-AD tauopathies, such as Pick bodies, astrocytic tau deposits, and dystrophic neurites. T807 did not bind brain tissue from Lewy body diseases or to TDP43 inclusions.
T807 phosphor screen autoradiography painted a similar picture. The characteristic black binding appeared strongly, as a thick band, in the expected cortical regions in AD, but not in controls or slices from the α-synuclein diseases. With this method, however, the non-AD tauopathies were puzzlingly negative.
Finally, Marquie used a beautiful autoradiography method where the T807-exposed slice of brain tissue is dipped into photographic emulsion, left overnight in the dark, and developed much like a Kodak photo. This allows the scientists to visualize T807/AV1451-labeled tissue at subcellular resolution. “With this method we see that T807 perfectly co-localized with tau-containing neurons and with dystrophic neurites,” Marquie told the audience. Like the standard autoradiography, this method, too, showed strong tracer binding to tau pathology in AD but not cerebral amyloid angiopathy, DLB, MSA or TDP-43. The high-resolution images of T807-labeled tangles prompted wide admiration at HAI. In toto, Marquie noted that tangles and dystrophic neurites appear to account for most of the in-vivo T807 signal.
Like Marquie, Val Lowe of the Mayo Clinic in Rochester, Minnesota, reported that T807/AV1451 binding matched up closely with tau PHF-1 but not with TDP-43 immunoreactivity in sections of human brain tissue. “We find exactly what Marta saw,” Lowe said. Lowe showed examples of FTD cases that were due to TDP-43 pathology, none of which bound T807/AV1451. Clinically, those were bvFTD or semantic dementia or PPA, and genetically, they were due to progranulin mutations or unknown causes, but in all cases their postmortem brain showed TDP-43 inclusions, not tau pathology.
[Images courtesy of Val Lowe, Mayo Clinic.]
Top left: Confirmed case of Alzheimer's. Autopsy picture (A) shows macroscopic slab of brain at the level of the amygdala (a). Microscopy sections of the boxed region show strong autoradiography signal with the tau tracer AV1451 (B). Tau antibody staining is strong in amygdala (C, arrow), showing pretangles, tangles, and neuritic threads at high magnification (E). This case had no TDP-43 (D, F.) .
Bottom: Confirmed case of FTLD-TDP. Autopsy picture (A) shows macroscopic slab of brain at the level of the amygdala (arrows in B-D). Microscopy sections of the boxed region show no autoradiography signal with AV1451 (B) or tau antibody staining (C at low, E high magnification). TDP-43 antibody staining was not discernible at low magnification (D) but revealed neuronal cytoplasmic inclusions and neuritic pathology at high magnification (F).
T807/AV1451 not binding TDP-43 inclusions was an important finding at HAI this year, and a relief to the field. Last year, unexpectedly positive T807/AV1451 scans in living patients whose symptoms were thought to be due to TDP-43 had raised the uncomfortable prospect that the new tracer might not distinguish those two major types of protein aggregation. These postmortem studies confirm that it does, said Keith Johnson of Massachusetts General Hospital.
Besides Marquie and Lowe, Yin-Guo Lin of Avid Radiopharmaceuticals of Philadelphia, working with researchers at Northwestern University in Chicago and the Banner Sun Health in Sun City, Arizona, reported similar findings. Lin saw no overlap between a fluorescent form of T807/AV1451 and TDP-43 immunohistochemistry. He did see strong autoradiography in areas rich in tau but not TDP-43 pathology. This postmortem validation study used 50 brain-slice sections of 30 cases with AD, various forms of FTD, and a normal control.
However, there are niggling problems with T807/AV1451, as well. They fall into two separate buckets: unexpected binding to brain regions that make no apparent sense versus absence of expected binding in tau-laden regions, and the behavior of the tracer in the brain during the course of the imaging session. The puzzling binding starts with the apparent absence of signal in some tauopathies such as Pick’s disease or corticobasal degeneration. Both Marquie and Lowe reported this. As explanations, Lowe suggested that T807/AV1451 might bind preferentially to 4-repeat tau over 3-repeat tau, and Marquie thought the tracer might recognize the paired helical filaments of AD more strongly than the straight helical filaments of CBD. At HAI, Dennis Dickson of the Mayo Clinic in Jacksonville, Florida, reviewed the bewildering variety of aggregated forms of tau in the spectrum of known tauopathies (watch his presentation here). PSP is of particular urgency to scientists, as several groups are planning therapy trials in this rare, pure tauopathy; alas, the first few cases being scanned with T807/AV1451 or studied postmortem have not yet generated a clear picture, and more research is needed.
The puzzling binding continues with off-target signals. Several groups using T807/AV1451 reported that it generates a fairly strong signal in parts of the brain’s basal ganglia, e.g., the striatum and substantia nigra, regardless of the patient’s diagnosis. In postmortem slices, Marquie saw this, too, yet she found no tau pathology in adjacent sections. Some groups see a thin line of what they believe is off-target binding at the edge of the hippocampus. T807/AV1451 also appears to bind the choroid plexus in scans. It is unclear where this binding comes from and what it means.
These early observations prompted extensive discussion at HAI. Scientists agreed that a definitive grip on the issue will require more postmortem and ultimately autopsy samples. In the meantime, Gil Rabinovici of the University of California, San Francisco, called on all groups to pool their data and contrast and compare jointly. Applauding this suggestion, Johnson said, “The main story here is that this tracer works well for AD. It shows a strong signal in the expected areas, now confirmed by autoradiography. FTDs are rare and more complex pathologically, so it will take longer to have enough cases to settle these questions.”
The second “bucket” of tracer issues that remain to be worked out concerns the tracer’s kinetics. At HAI, Suzanne Baker of Lawrence Berkeley National Laboratory in Berkeley, California, took a crack at characterizing how T807/AV1451 behaves during the course of a long scanning session. “With PiB a lot of modeling and careful, quantitative in vivo work has been done, but with the tau tracers people are so excited to get them into trials that this technical work lags behind. It is important to do that if you want to measure small changes over time or in response to a drug, especially in a multicenter trial,” she told Alzforum.
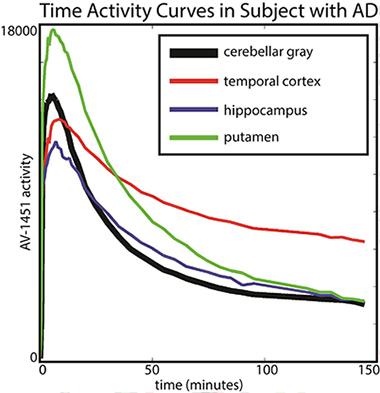
Baker scanned 19 older healthy controls and 14 people with AD for 150 minutes (with a 20-minute break before the last half-hour) and analyzed how the tracer distributed between different brain areas and how that changed over time. Her key finding was that in AD patients, different brain regions had different kinetics. In particular, T807/AV1451 in the cortex did not reach a steady state, i.e., a window of time during which the ratio of binding in a target region (i.e. temporal cortex) to binding in the reference tissue (i.e. cerebellum) was stable. This is the most robust time frame for imaging because scientists will get a reproducible measurement from scan to scan. Instead, the kinetics of T807/AV1451 turned out to be fast in subcortical regions where the tracer is thought to have some off-target binding, intermediate in other regions, and so slow in the cortical regions—perhaps because they contain the most tau—that their uptake never stabilized in the 150-minute scanning period. “We were astonished by the variability across regions that we saw,” Baker said.
The putamen shows faster kinetics and the temporal cortex shows slower kinetics in comparison to cerebellar gray (reference region). [Image courtesy of Suzanne Baker and William Jagust.]
This finding essentially implies that the tracer’s uptake is a moving target. For example, a scan taken between 80 and 100 minutes after injection might give a different result than one taken between 120 and 140 minutes after injection, raising questions about how best to compare tracer uptake between different brain regions and in longitudinal studies.
These uncertainties do not question the tracer’s ability to detect whether there is a significant amount of tau pathology in a given brain region, only the finer quantitative measurements, Baker said. The problems require careful attention to issues of timing of scan acquisition.
Baker’s talk prompted extensive discussion and some jokes about 12-step programs. “The first step is admitting you have a problem,” she quipped. Others agreed that more kinetic modeling can help solve it, ideally with a study that uses arterial input data to better approximate how much tracer enters the brain at which time point after injection. Some kinetic modeling studies are underway at Avid Radiopharmaceuticals and at MGH.
In summary, Chet Mathis of the University of Pittsburgh recommended that all tau tracers that will be used clinically be subjected to both careful ex vivo validation and pharmacokinetic modeling to clarify their specific binding and distribution between regions and compartments in the brain. “We have to do the hard work before we can interpret the scans,” Mathis said.
On the other hand, PET experts also largely subscribe to the notion embodied by a quote by former Defense Secretary Donald Rumsfeld: “You go to war with the army you have … not the army you might want or wish to have at a later time.” For FDA approval, PET tracers will most likely need autopsy correlations, which have started but take a long time to obtain. Scientists are beginning to suspect that tau’s close correlation to neurodegeneration and clinical symptoms means tau PET might prove to be a good outcome marker in therapy trials, whereas amyloid PET might be more useful as an inclusion criteria at screening, and they are eager to test whether this is true. “Tau PET measures a molecular pathology, and it is close to the action. That combination has been missing,” Johnson told Alzforum.
Bill Klunk of UPitt, who co-developed PiB with Mathis, emphasized that none of the technical questions with T807/AV1451 are show-stoppers. “Let’s not throw the baby out with the bathwater. Let’s just clean up the bathwater. When you look at the overall results with T807 so far, it’s clear the baby is still really good,” Klunk said.—Gabrielle Strobel

Comments
No Available Comments
Make a Comment
To make a comment you must login or register.