Natural History Studies Provide Foundation for FTD Research
Quick Links
At the 11th International Congress on Frontotemporal Dementia, held November 11–14 in Sydney, no two acronyms were heard more than ARTFL and LEFFTDS. For those readers who have not built them into their FTD vocabulary yet, Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) and Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) are closely related, ongoing longitudinal studies of familial and, in the case of ARTFL, also sporadic FTD. Researchers are banking that these two cohorts, and their proposed continuation under the umbrella of a single study called ALLFTD, a.k.a. the ARTFL LEFFTDS Longitudinal FTD study, will chart onset and progression of the main types of FTD, and in the process build well-characterized, trial-ready cohorts for future therapeutic studies. ARTFL and LEFFTDS began in 2014, garnering the biggest grant the NIH ever gave to FTD research (Nov 2014 conference news). Where do they stand now?
- ARTFL and LEFFTDS have recruited 1,139 and 367 volunteers, respectively.
- Longitudinal analysis has begun to discern patterns among FTD subtypes.
- Researchers hope to continue both projects under a new ALLFTD grant.
Leah Forsberg, Mayo Clinic, Rochester, Minnesota, reviewed LEFFTDS, while Adam Boxer of the University of California, San Francisco, laid out ARTFL progress. Bradley Boeve, also at the Mayo in Rochester, and Howard Rosen of UCSF co-lead the NIH-funded LEFFTDS, while, as part of the larger Rare Diseases Network, the ARTFL consortium is coordinated by Rosen, Boxer, Hilary Heuer, and others at UCSF. Both studies share a similar design and even subjects, with all but four LEFFTDS participant also enrolled in the ARTFL familial FTD cohort, Heuer told Alzforum. The two studies collect the same types of biological data and store it in a common database. They both aim to track rates of decline and look for genetic variants and biomarkers that modulate disease onset and progression.
Forsberg reported that seven centers in the U.S. and one in Canada had recruited 367 volunteers from 65 families into LEFFTDs as of late October. Almost all are Caucasian, seven are Asian, and 10 represent all other non-Caucasians. Fifty-five percent are women. Volunteers range from 35 to 63 years old and have about 15 years of education on average. About 129 healthy participants carry none of the known FTD mutations and show no signs of FTD. About the same number carry a mutation and are asymptomatic, while 110 have a mutation and are asymptomatic.

Clinical Diagnoses. The range of diagnoses among ARTFL volunteers with sporadic FTD. [Courtesy of Adam Boxer.]
LEFFTDS had hoped to recruit equal numbers of people from families carrying mutations in the tau gene MAPT, the progranulin gene GRN, or expansions in the C9ORF72 gene. They pretty much succeeded, recruiting 26, 28, and 33 percent of the cohort from such families, respectively. Ten distinct MAPT and 18 distinct GRN mutations are represented in the cohort. Of 145 people from families carrying C9ORF72 hexanucleotide expansions, two people had both an expansion and a progranulin mutation. Forsberg said that because the genotype data is de-identified, LEFFTDS researchers do not know an individual’s mutation status unless that person reveals it.
To date, all participants have had one clinical visit. Seventy percent have already had a second, 52 percent have had a third, and 16 percent a fourth. About 90 percent of participants have volunteered DNA, RNA, and plasma/serum; have undergone MRI imaging; and have donated peripheral blood mononuclear cells, which can be used to make induced pluripotent stem cells to study the effects of the various mutations on cell biology. Only about 45 percent have agreed to donate cerebrospinal fluid; this is not unusual among North Americans who seem more anxious about lumbar punctures than Europeans. All were evaluated clinically and took a battery of neuropsychological tests.
For its part, ARTFL has recruited 608 people who have sporadic FTD. Among them, 184 had behavioral variant FTD, 128 with progressive supranuclear palsy, 76 with corticobasal syndrome, and 77 and 62 with semantic and non-fluent variants of primary progressive aphasia (see image above). Eighteen had FTD-ALS. As in LEFFTDS, participants were predominantly Caucasian, with a similar number of men and women except for the bvFTD and FTD-ALS patients, of whom 72 and 61 percent, respectively, were men. Of the 608, researchers concluded that 62 did not have FTD but another disease, such as Alzheimer’s, Parkinson’s, or dementia with Lewy bodies.

ARTFL Diagnoses. The breakdown of clinical diagnoses among ARTFL FTD mutations carriers. [Courtesy of Adam Boxer.]
In addition to the 608 people with sporadic FTD, 531 people from familial FTD kindreds have also enrolled in ARTFL. These are families who carry a known FTD mutation, including 363 of the people enrolled in LEFFTDS, or who report a family history that indicates an autosomal-dominant cause. Ninety-two people fell into the latter category, while 184, 104, and 132, came from C9ORF72, GRN, and MAPT families, respectively. Only 19 people had other variants, including those in the VCP and TARDBP loci. Among the 531 recruits, 333 show no symptoms, 84 have mild symptoms, and 114 have full-blown disease, meaning a CDR of 1.0 or greater. Like LEFFTDS, they are predominantly Caucasian and 56 percent are women. Ages ranges from 31 to 70.
ARTFL collects the same biological samples as does LEFFTDS, except no CSF. Among ARTFL volunteers with fFTD, 317 have completed a one-year follow-up visit. Most showed no change in clinical status; 16 went from asymptomatic to mildly impaired. Of those who had symptoms at baseline, seven seemed to improve, meaning they went from CDR 0.5 to CDR 0, while 23 worsened.
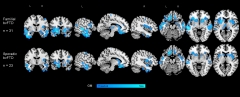
Different Cause—Same Pattern. Compared with 30 healthy controls (not shown), people with familial and sporadic bvFTD show similar brain atrophy patterns. [Courtesy of Adam Boxer.]
Many of the participants in ARTFL/LEFFTDS have not been seen often enough for researchers to reach consensus about deterioration among the various subtypes of FTD; still, some trends are beginning to emerge. On a poster, Boxer directly compared sporadic and familial behavioral variant FTD in the ARTFL cohort. He reported that people with MAPT mutations were younger at diagnosis than those with any other mutation or with sporadic bvFTD. This might imply MAPT mutations cause a more aggressive disease, yet Boxer saw little difference in prevalence of neuropsychiatric symptoms among the volunteers. If anything, those with sporadic bvFTD were more likely to be reported by family members or carers as being depressed and irritable. Clinicians also reported more irritability among sporadic bvFTD patients as disease progressed, but found no neuropsychiatric differences between familial and sporadic bvFTD on the first visit to the clinic. MRI data supports the idea that sporadic and familial bvFTD are similar. Atrophy patterns among 23 sporadic and 31 familial patients were all but indistinguishable (see image above), at least for this behavioral variant form. This might be good news for clinical trials, because it suggests that the patients could be combined, making it slightly easier for clinicians to recruit people with this rare disease. For more on outcome measures that might be used in such trials, and on heterogeneity of atrophy patterns among FTD patients in general, see Part 3 of this series.
Going forward, ARTFL/LEFFTDS researchers hope to follow all these patients for at least another five years under the auspices of the ALLFTD study. Rosen told Alzforum that ALLFTD will recruit new participants also. ALLFTD plans to test the predictive value of polygenic risk scores in addition to biomarkers. “We are taking advantage of new research developments and hope to incorporate other data, such as whole genome sequencing, through collaboration with other efforts,” Rosen said.—Tom Fagan
References
News Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.