A Proteomics Dive into Cause of Frontotemporal Dementia
Quick Links
If DNA makes RNA makes protein, then genomic variants make RNA make protein … make disease. While scientists have found polymorphisms that associate with various forms of frontotemporal dementia, deciphering how they perturb proteins is proving more complicated. “It’s time we start thinking how to get beyond genetics to a functional understanding of FTD,” said Raffaele Ferrari, University College London, at the 11th International Congress on Frontotemporal Dementia, held November 11–14 in Sydney. Ferrari and others championed the idea that studying co-expression patterns and protein-protein interactions will not only lay bare the underlying biology of FTD, but also identify disease markers and therapeutic targets.
- Scientists are beginning to uncover protein interactions underlying FTD.
- Lysosomal and immune functions stand out as top hits.
- Immunotherapies are being tested to counter dysfunctional pathways.
At the conference, some researchers described functional protein modules related to progranulin insufficiency that suggest early inflammatory and lysosomal changes in FTD. Mutations in the progranulin gene are a major cause of familial FTD, while levels of the protein drop in people with sporadic forms as well. Others showed that progranulin-related changes can be cell-specific, tracking one way in microglia and the opposite way in whole brain. They also made a much-sought connection between two key players in the pathology of FTD and amyotrophic lateral sclerosis (ALS)—the lysosome and RNA granules. Last but not least, scientists from Alector, a small startup in South San Francisco, reported that their therapeutic antibody targeting sortilin, a receptor that promotes lysosomal degradation of progranulin, boosts levels of the protein in mice and monkeys and rescues behavioral deficits in the former. AL001 has entered clinical trials.
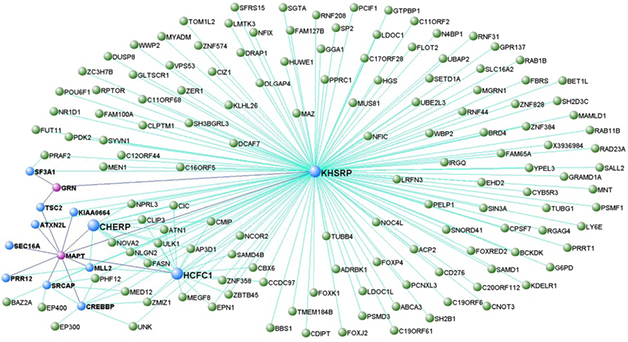
FTD Modules. A representative model from co-expression analysis places genes for tau (MAPT) and progranulin (GRN), in a network that regulates DNA transcription. MAPT and GRN are colored pink, while genes directly connected to them are blue. In this module, MAPT was a hub. [Courtesy of Ferrari et al., Molecular Neurodegeneration.]
Modules and Cells
For his part, Ferrari capitalized on data from the International FTD Genomics Consortium in his search of gene network changes that point to molecular pathways gone awry in FTD. Using transcription microarray analysis of frontal and temporal cortices from 101 samples from the U.K. Human Brain Expression Consortium, Ferrari uncovered networks of genes that co-express with known FTD genes, including MAPT, GRN, C9ORF72, VCP, OPTN, and TMEM106B. These co-expression analyses turned up modules of genes involved in DNA protection and transcriptional regulation, immune function, lysosomal activity, proteostasis, and the ubiquitin proteasome pathway (Ferrari et al., 2016).
How do these modules function? For that, Ferrari turned to proteomics. He devised a layered approach to build protein interactomes. First, he used products of FTD genes, such as tau and progranulin, as seeds in a weighted protein-protein interaction network analysis. He then used nodes from those protein networks as seeds in subsequent analysis (Ferrari et al., 2017). Putting it all together, he ultimately identified blocks of proteins linked to FTD genes that are involved in DNA repair, cytokine-mediated immune responses, the ubiquitin proteasome system, and the unfolded protein response. “We thought it quite supportive, from disease-mechanism and functional perspectives, that the protein-protein interaction and expression networks, which are both based on Mendelian and GWAS hits, substantially overlaid each other,” he said. The overlap was highest for DNA repair and cellular waste disposal. Among these were four interactome hubs, which were defined as proteins common to at least eight interactomes. One was EP300, a histone acetyltransferase that Ferrari considers a potential biomarker or therapeutic target.
At the conference, others asked if this analysis can pick out what wayward pathway or molecule kick-starts the disease process. Ferrari thinks not. “The more pressing point is that we have done this work in silico, so we need to validate experimentally,” Ferrari said. “We have some of that data because the protein-protein interactions have been determined biochemically, but still need further and more tailored work in FTD models.”
Scientists are using animal models to identify the protein networks and interactomes germane to FTD. The labs of Thomas Kukar, Nicholas Seyfried, and Malu Tansey at Emory University, Atlanta, have compared the brain proteomes of progranulin knockout mice and people with progranulin mutations to find common pathways that might be pathogenic. In the mice, a deep proteome analysis quantified 8,000 different proteins and found differences between Grn KOs and wild-type controls, said Kukar. In three-month-old knockouts, 29 proteins were upregulated over age-matched wild-type, and 26 were downregulated. The proteins included cathepsins B, D, and Z, TPP1, HexA and B, and glial proteins such as GFAP and complement C1q, pointing to lysosomal dysfunction and glial activation. “Lysosome dysregulation and inflammation have been linked to FTD before, but these mice are only three months old, so we think such changes occur earlier than expected and may serve as novel biomarkers of disease progression,” said Kukar. To his mind, this also suggests a subtle disconnect between proteome and transcriptome changes, since the latter are not seen in such young animals. This hints that disease might start with protein-level changes that then cause transcriptional dysregulation.
In 19-month-old mice, the number of upregulated proteins had grown to 119, while the number of downregulated proteins was about the same as in the younger mice. The same lysosomal and microglial proteins were still differentially expressed at that age, but so were new glial markers—complement C4 and S100a4.
Since FTD GRN mutations are heterozygous, how would these changes lead to human disease? By immunohistochemistry, Kukar showed that Grn-heterozygous mice had upregulated the lysosomal activation CD68 by 12 months, and even more by 24 months of age. “We think lysosomes are dysfunctional in Grn heterozygotes, they just need to be older than full knockouts for changes to manifest,” said Kukar.
Like Ferrari, Kukar analyzes correlation networks to probe functional consequences of proteome changes. He identified 29 protein modules in mice. Four most strongly correlated with knockout status and age. To relate this to FTD, he compared the mouse modules with human ones identified in frontal cortex samples from 11 age-matched controls, seven GRN-FTLD cases, and 10 non-GRN cases (Chen-Plotkin et al., 2008). He found 10 modules that overlapped between the mouse Grn knockouts and the familial and sporadic FTD cases.
In Sydney, Kukar focused on a common human/mouse module that was enriched in microglial proteins, complement factors, and proteins found in peripheral immune cells. “This might be evidence that peripheral cells have entered the brain in FTD,” he said, adding that the Tansey lab is now using flow cytometry to measure how many microglia and peripheral immune cells there are in the brain samples. In mice, flow cytometry indicates older Grn knockouts have fewer microglia in the brain than do age-matched wild-type animals. “This was surprising, because the literature would suggest they should have more,” Kukar said. It turns out that peripheral monocytes might make up for the shortfall. Based on cell-surface expression of CD45 and CD11b, the scientists found a strong infiltration of monocytes into Grn knockout brain. Whether the same happens in FTD remains to be seen. As a start, Tansey and colleagues are measuring changes in peripheral blood monocytes from volunteers in the ARTFL/LEFFTDS natural history studies. The data could have important implications for therapeutic strategies in FTD caused by GRN haploinsufficiency, Kukar said.
Looking to Lysosomes
This idea of changes to immune cells in FTD drew further support from Anja Capell, Ludwig Maximilians-University, Munich. Capell also uses mice, but studies directly how deleting GRN affects lysosomal function. Previously, she reported that in 22-month-old mice and mouse embryonic fibroblasts (MEFs), progranulin deficiency results in higher expression of the lysosomal proteins cathepsin B and D, LAMP1, and saposin D. This led to more maturation and activation of cathepsins, which are initially made in an inactive prepro form (Götzl et al., 2014).

Cell-Specific Responses.
Without progranulin, lysosome activity (yellow-red color scale) increases with age in neurons (green), perhaps to compensate for faltering lysosomes in microglia (blue). [Courtesy of Anja Capell.]
Does this cathepsin boost square with lysosome dysfunction in FTD? After all, the lysosome is there to degrade proteins. To investigate, Capell and colleagues used S35 pulse labelling to check protein degradation in MEFs. Indeed, MEFs from GRN knockouts degraded lysosomal substrates, in this case the amyloid precursor protein and its C-terminal fragments, faster than did wild-type MEFs. Capell also saw an age-related uptick in expression of lysosomal membrane proteins such as Cd68 and Ctsd in GRN knockout brain, again pointing to up-, not down-, regulation of lysosomes.
Things started to make sense when the researchers specifically examined microglia they captured from brain extracts using microbeads coated with antibodies to the microglial cell surface marker CD11b. In contrast to whole-brain extracts from GRN knockouts, the microglia did downregulate expression of cathepsin D, and suppressed maturation of cathepsin B. In short, Capell believes that without progranulin, lysosomes become dysfunctional, particularly in microglia, and that this leads to enhanced lysosomal activity in other brain cells.
Others wondered why microglia seem to be persistently activated in older GRN knockouts. Capell suggested this might be the brain’s attempt to compensate for reduced lysosomal activity.
Taking yet another proteomic tack, Michael Ward from the National Institutes of Neurological Disorders and Stroke, Bethesda, Maryland, forged a long-anticipated connection between lysosomes and RNA granules. The latter are hotbeds for accumulation and, potentially, aggregation of RNA-binding proteins that build up in neurons in FTD and ALS, such as TDP-43, FUS, and hnRNPA1. As detailed in a poster, Ward and colleagues, including co-first authors Michael Fernandopulle in his lab, and Ya-Cheng Liao in Jennifer Lippincott-Schwartz’s group at the Howard Hughes Medical Institute, Ashburn, Virginia, used proximity-labelling proteomics to identify proteins that bind to lysosomes. The scientists tied those organelles to axonal transport of RNA granules, which may go awry in ALD/FTD. Meeting organizers awarded the poster first prize in the category for investigators who are early in their career.
In proximity labelling, researchers engineer a protein, lysosomal LAMP1 in this case, to incorporate an ascorbate peroxidase (APEX) unit (Rhee et al., 2013). Add a little hydrogen peroxide and biotin to cells expressing LAMP1-APEX, and presto, it biotinylates nearby proteins, which can then be tracked or isolated using streptavidin. In this manner, Fernandopulle identified 99 different proteins that interact with lysosomes in neurons derived from patient-induced pluripotent stem cells. Comparing those with hits from proximity-labelling of RNA stress granules reported earlier this year by researchers in Gene Yeo’s lab at University of California, San Diego (Markmiller et al., 2018), Fernandopulle found six proteins in common. One was none other than annexin A11 (ANXA11), a vesicular-trafficking protein that is mutated in some ALS patients (May 2017 news; Liu et al., 2018; Tsai et al., 2018).
Ward explained that RNA stress granules use ANXA11 to hitch a ride on endolysosomes as they move through axons. ANXA11 contains a low-complexity N-terminal domain predicted to undergo liquid-liquid phase separation, suggesting the protein blends into the liquid droplets that constitute RNA granules. Its C-terminal contains repeats that interact with membrane lipids and calcium. In collaboration with Peter St George Hyslop’s group in Cambridge, England, Ward showed that ANXA11 undergoes liquid-liquid phase separation and that it interacts with both RNP stress granules and lysosomes in living cells. The ALS-linked ANXA11 mutations D40G, R235Q, and R346C all disrupted phase separation of stress granules and reduced lysosomal binding.
Using a light-activated form of ANXA11 and time-lapse photography, Liao showed that annexin A11 brings lysosomes and RNP granules together and that they travel as one in axons of rat neurons. Again, ALS mutations scupper this interaction and reduce co-trafficking of RNA granules and lysosomes. All told, the work ties RNA granules to lysosomes via ANXA11, and suggests that damage to this interaction contributes to the pathophysiology of both ALS and FTD, said Ward.
A Lysosome-Based Immunotherapy
Showing how the study of protein-protein interactions can pay dividends, Michael Kurnellas updated the audience on Alector’s anti-sortilin program. The company targeted sortilin-1 because mice lacking this sorting receptor produce more progranulin and have no obvious behavioral or lysosomal phenotype (Nov 2010 news on Hu et al., 2010). The researchers developed aSORT1, a.k.a AL101, a monoclonal antibody that binds sortilin-1 with high affinity and prevents progranulin from docking with the receptor. In both wild-type and GRN+/- heterozygous mice, the antibody all but abolished sortilin-1 in white blood cells, while boosting plasma and CSF levels of progranulin about six- and twofold, respectively. Kurnellas did not disclose how much antibody was administered, or for how long. He did report that the same antibody slashes levels of sortilin in cells from cynomolgus monkeys, while boosting serum and CSF levels three- and twofold, respectively.
Does this help? To find out, Kurnellas and colleagues collaborated with Erik Roberson at the University of Alabama, Birmingham. Andrew Arrant in the lab had reported that progranulin-heterozygous mice are a bit of a pushover when they meet another mouse face-to-face in a small tube. They are much more likely to retreat. However, a weekly regimen of aSORT1 made 21-month-old heterozygotes more likely to hold their ground than those treated with a control IgG antibody.
This past August, the U.S. Food a,nd Drug Administration granted orphan drug status to AL001, a humanized monoclonal version of aSORT1. INFRONT, a single-ascending-dose Phase 1 trial began in September. It is slated to recruit 60 healthy volunteers at seven sites in North America and Europe. Kurnellas said a multidose trial in FTD patients carrying progranulin mutations will follow once Phase 1 has completed. INFRONT is expected to finish by March 2020.—Tom Fagan
References
News Citations
- Potential New ALS Gene Leads to Extraordinary Aggregates
- Sorting Progranulin With Sortilin—New Clues to FTLD Pathology
Therapeutics Citations
Paper Citations
- Ferrari R, Forabosco P, Vandrovcova J, Botía JA, Guelfi S, Warren JD, UK Brain Expression Consortium (UKBEC), Momeni P, Weale ME, Ryten M, Hardy J. Frontotemporal dementia: insights into the biological underpinnings of disease through gene co-expression network analysis. Mol Neurodegener. 2016 Feb 24;11:21. PubMed.
- Ferrari R, Lovering RC, Hardy J, Lewis PA, Manzoni C. Weighted Protein Interaction Network Analysis of Frontotemporal Dementia. J Proteome Res. 2017 Feb 3;16(2):999-1013. Epub 2017 Jan 12 PubMed.
- Chen-Plotkin AS, Geser F, Plotkin JB, Clark CM, Kwong LK, Yuan W, Grossman M, Van Deerlin VM, Trojanowski JQ, Lee VM. Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Hum Mol Genet. 2008 May 15;17(10):1349-62. PubMed.
- Götzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, Janssens J, van der Zee J, Lang CM, Kremmer E, Martin JJ, Engelborghs S, Kretzschmar HA, Arzberger T, Van Broeckhoven C, Haass C, Capell A. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 2014 Mar 12; PubMed.
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013 Mar 15;339(6125):1328-1331. Epub 2013 Jan 31 PubMed.
- Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo EC, Krach F, Yang D, Sen A, Fulzele A, Wozniak JM, Gonzalez DJ, Kankel MW, Gao FB, Bennett EJ, Lécuyer E, Yeo GW. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell. 2018 Jan 25;172(3):590-604.e13. PubMed.
- Liu X, Wu C, He J, Zhang N, Fan D. Two rare variants of the ANXA11 gene identified in Chinese patients with amyotrophic lateral sclerosis. Neurobiol Aging. 2019 Feb;74:235.e9-235.e12. Epub 2018 Sep 22 PubMed.
- Tsai PC, Liao YC, Jih KY, Soong BW, Lin KP, Lee YC. Genetic analysis of ANXA11 variants in a Han Chinese cohort with amyotrophic lateral sclerosis in Taiwan. Neurobiol Aging. 2018 Dec;72:188.e1-188.e2. Epub 2018 Jul 10 PubMed.
- Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010 Nov 18;68(4):654-67. PubMed.
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.