TREM2, Microglia Dampen Dangerous Liaisons Between Aβ and Tau
Quick Links
That microglia play a central role in neurodegenerative disease has become a premise; it’s no longer a question. The questions are all about the how. This was abundantly clear at joint symposia held June 16–21 in Keystone, Colorado. At Neurodegenerative Diseases: New Insights and Therapeutic Opportunities and Neural Environment in Disease: Glial Responses and Neuroinflammation, scientists exchanged their latest insights on how these exquisitely reactive glial cells shape the course of pathology, inflammation, and neurodegeneration. Via their surface receptor TREM2, microglia reportedly sculpt the architecture of Aβ plaques to lessen the formation of dystrophic neurites. Left to develop unchecked, these deformed neuronal extensions serve as fertile breeding grounds for tau aggregates, scientists found. Other researchers presented initial data from a chimeric AD mouse model flush with human microglia.
- TREM2 facilitated denser plaques, fewer dystrophic neurites.
- Dystrophic neurites seed tau aggregation and propagation.
- Depletion of microglia prevented plaque formation.
The preponderance of microglial genes among AD risk variants place these cells front and center in the disease. TREM2, the strongest among them, has been implicated in all manner of microglial functions. At Keystone, David Holtzman of Washington University in St. Louis placed TREM2 at the intersection between Aβ and tau pathology. As presented last year at Keystone by collaborator Christian Haass of the German Center for Neurodegenerative Diseases in Munich, and subsequently published, the scientists reported that without TREM2, mice were unable to rally microglia around Aβ plaques. This resulted in fluffier, more diffuse plaques infused with about half as much ApoE as those in mice replete with TREM2 (Jul 2018 conference news; Jan 2019 news). Holtzman also previously reported that in the PS19 model, which express the P301S mutant form of tau that causes frontotemporal dementia, TREM2 deficiency quelled harmful neuroinflammatory responses (Oct 2017 news).
This year, Holtzman put the two together, reporting how the microglia-plaque relationship affected the development of tau pathology. A graduate student in the lab, Cheryl Leyns, who is now at Merck, injected tau-laden material extracted from Alzheimer’s brains into the brains of 3-month-old APP/PS1 mice. A month later, she observed tau aggregates mingling with dystrophic neurites surrounding Aβ plaques, at least on the same side of the brain where she had made the injection.
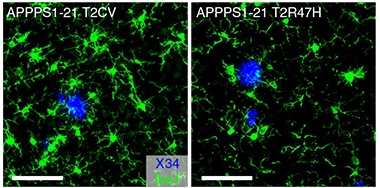
Sluggish Recruitment. Microglia (green) rallied enthusiastically around plaques (blue) in mice expressing the common variant of TREM2 (left), but not the R47H variant (right). [Courtesy of Leyns et al., Nature Neuroscience, 2019.]
However, in APP/PS1 mice lacking Trem2, these dystrophic neurite-associated tau aggregates doubled. They even cropped up on the other side of the brain. A similar doubling of tau aggregates also occurred in mice expressing the R47H mutant form of human TREM2. These tau aggregates contained only endogenous mouse tau, suggesting they had formed by way of templated misfolding triggered by the injected human AD-tau.
Notably, the amount of aggregated tau correlated with the burden of dystrophic neurites in the brain, which, in turn, was higher in mice lacking functional TREM2. No tau aggregates had appeared when the researchers injected AD-tau before dystrophic neurites were forming, casting the damaged processes as a fertile substrate for subsequent seeding of tau. The findings appeared in Nature Neuroscience on June 24 (Leyns et al., 2019).
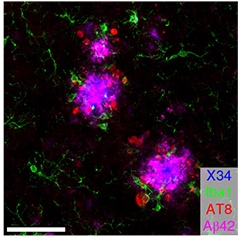
Sowing the Seeds of Tau. In a TREM2 knockout APP/PS1 mouse injected with AD-tau, tau aggregates (red) cluster around Ab42-laden (pink) plaques (blue). Microglia (green) hardly budge. [Courtesy of Leyns et al., Nature Neuroscience, 2019.]
Holtzman speculated that microglia tweak the architecture of plaques in a manner that requires TREM2 to function. His findings mesh with previous work led by Jaime Grutzendler of Yale University in New Haven, Connecticut. Grutzendler reported that without TREM2, microglia have trouble erecting a barrier around plaques, leading to “hotspots” of Aβ42 that wreak havoc on nearby axons (May 2016 news). Holtzman speculated that toxic tau could sprout from the ruins. “Dystrophic neurites, which are full of abnormal membranous material, may be a good substrate for allowing tau to convert to this irregular conformation,” he told Alzforum.
Haass believes that while this did not prove a connection between the two hallmark proteinopathies of AD, the results suggest as much. “Maybe the TREM2-dependent microglial function is the missing link that connects amyloid and tau pathologies,” he said.
Also at Keystone, David Hansen of Genentech in South San Francisco presented data that dovetailed with Holtzman’s. Hansen reported that, compared with PS2APP mice expressing TREM2, PS2APP mice lacking TREM2 had fluffier plaques and a higher proportion of Aβ42 to Aβ40. Hansen also reported an increase in LAMP1-positive, i.e. lysosome-packed, dystrophic neurites per plaque in TREM2 knockouts, and even found these damaged processes far away from plaques. Hansen’s group further examined structural tissue damage at the level of dendritic spines, reporting that TREM2 deficiency worsened spine loss near plaques.
What does all this mean for tau pathology? Hansen studied this in TauPS2APP model mice, which are PS2APP mice that express the P301L mutant form of tau. By nine months of age, TauPS2APP mice crossed to a TREM2-deficient background had more tau pathology than mice that did express TREM2.
Overall, Hansen drew similar conclusions to Holtzman: “Microglia, through TREM2, mitigate the degree to which Aβ pathology accelerates tau pathology,” he told the audience. Leyns, too, noted that together, both groups’ findings suggest that without TREM2, plaques assume a more Aβ42-laden form that damages neurons and creates a permissive environment for tau seeding.
Kim Green of the University of California, Irvine, reported that microglia not only compact plaques, but are required to build them in the first place. At Keystone, Green described what happened when he treated 6-week-old 5xFAD mice, which have no plaques yet, with the CSF-1R inhibitor PLX-5622. The treatment, which Green developed, depletes the mice’s microglia down to 2 percent of their normal numbers within a few days. Surprisingly to Green, as long as the mice’s chow contained PLX-5622 to keep microglia at bay, the mice developed barely any plaques. At 4 months of age, the few plaques that had grown were surrounded by those remaining few microglia.
Green fed PLX-5622 to the mice for as long as six months, eventually wiping out all their microglia. After that course, some plaques remained in the same regions where microglia had initially persisted with the shorter treatments, hinting the cells had been instrumental in building those plaques. Without microglia to compact them any longer, these remaining plaques had morphed into diffuse, fluffy deposits surrounded by dystrophic neurites, similar to those Holtzman observed. Green also reported that when PLX-5622 was initiated after plaques had formed, it did not affect overall plaque numbers.
Holtzman noted that while microglial depletion prevented plaques from forming in Green’s experiments, TREM2 deficiency merely altered the plaques’ architecture without strongly changing their overall burden in both his and Hansen’s work. This implies microglial factors other than TREM2 help build plaques, he said.
Green further reported that after six months on the inhibitor, the 5xFAD mice had large amounts of Aβ aggregate on the walls of their cerebral blood vessels, akin to cerebral amyloid angiopathy (CAA). Green suspects that without microglia corralling Aβ in the parenchyma, Aβ gets shunted into the vasculature instead. When the scientists stopped the inhibitor treatment and waited a month, microglia returned and plaques formed, though the CAA remained.
Green found no cognitive side effects of prolonged treatment with this drug, though some researchers wondered whether the CAA could cause cognitive deficits.
Green emphasized that he does not advocate wiping out microglia as a treatment in humans. After all, the cells have beneficial functions such as compacting plaques and limiting their neurotoxicity. However, he noted that in some scenarios, temporarily ridding the brain of stubbornly reactive microglia could theoretically allow new, less-damaging cells to repopulate it.
For example, a recent study reported that removal of reactive microglia, using PLX-5622, prevented “chemofog” triggered by methotrexate in mice. In that scenario, microglia riled up by the chemo drug derailed the development of oligodendrocytes, which led to deficits in myelination (May 2019 news). In another recent report, a necroptotic die-off of reactive microglia boosted remyelination in a model of multiple sclerosis (Jun 2019 news).
How fast do microglia rebound after PLX-5622 is gone, and where do the new cells come from? Monique Mendes, a graduate student in Anna Majewska’s lab at the University of Rochester in New York, addressed this question at Keystone. Using cranial windows to peer into the mouse brain before and after microglial depletion, Mendes saw that the few microglia that survived this treatment were the main suppliers of newborn microglia once the inhibitor was no longer there. Between two and four days after the mice stopped taking the drug, Mendes spotted microglia with two somas, some of which ultimately split into two cells. Only some microglia appeared destined for rapid division. While some microglia divided at a blistering pace—turning over within four to 12 hours per division—others remained trapped in a binucleate stage, apparently hesitant to split into two independent cells.
This data do not address whether microglia born into neurodegenerative environs might take on the reactive stance of their forebears, but Green told Alzforum that the finding jibes with a rapid repopulation his lab has observed as well. According to Mendes’ data, depleting 100 percent of microglia would preclude their recovery after treatment stops. Green told Alzforum that he will soon share data about what happens when every last microglia is depleted from the brain.—Jessica Shugart
References
News Citations
- TREM2: Diehard Microglial Supporter, Consequences Be DAMed
- Without TREM2, Plaques Grow Fast in Mice, Have Less ApoE
- Changing With the Times: Disease Stage Alters TREM2 Effect on Tau
- Barrier Function: TREM2 Helps Microglia to Compact Amyloid Plaques
- Microglia Instigate ‘Chemofog’ by Squelching Myelination
- Dead Microglia Pave the Way for Myelin Regeneration
Research Models Citations
Paper Citations
- Leyns CE, Gratuze M, Narasimhan S, Jain N, Koscal LJ, Jiang H, Manis M, Colonna M, Lee VM, Ulrich JD, Holtzman DM. TREM2 function impedes tau seeding in neuritic plaques. Nat Neurosci. 2019 Aug;22(8):1217-1222. Epub 2019 Jun 24 PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.