Cloak and Dagger Clusters? How C9ORF72 Repeats Kill Is Still a Mystery
Quick Links
When it first broke, the news that expanded sequences in the C9ORF72 gene are a major cause of both frontotemporal dementia and amyotrophic lateral sclerosis shook up both research communities (see Sep 2011 news story and Sep 2012 conference coverage). Three years later, research on this mutation has exploded, and was on full display at the 9th International Conference on Frontotemporal Dementias held October 23 to 25 in Vancouver, Canada. Researchers have spent this time generating tools to study the problem and are now ready to grapple with the questions that preoccupy them at this stage: How do the repeats wreak havoc on the nervous system? And why do some people with the expansions develop FTD, others ALS, and still others a combination of the two?
Tucked into an intron in the C9ORF72 gene, the simple sequence GGGGCC is harmless in small doses. Normal people have up to a couple of dozen repeats of the sequence, but people with ALS or FTD can harbor hundreds to thousands of sequential copies. Researchers estimate the run-on repeat accounts for 40 percent of familial ALS and about a quarter of familial FTD, as well as a smaller fraction of sporadic disease (see Majounie et al., 2012).
Once transcribed, the repeat sequences form secondary RNA structures called G-quadruplexes, which bundle up with RNA-binding proteins in nuclear aggregates called RNA foci. The repeats are intronic, yet they are translated in both the sense and antisense directions by a process called repeat-associated non-ATG, or RAN, translation. Depending on where in the mRNA translation starts and in what direction it moves, five different peptides can be produced, each made up of strings of just two amino acids: glycine-alanine (GA), glycine-proline (GP), glycine-arginine (GR), proline-alanine (PA), or proline-arginine (PR). These “dipeptide repeats,” or DPRs, have been reported to form intraneuronal inclusions (see Feb 2013 news story and Feb 2013 news story).
At the meeting, researchers proposed three possible ways in which C9ORF72 expansions might spell trouble for neurons. The sequestration of C9ORF72 RNA into foci has been reported to prevent its expression, although researchers do not yet know what C9ORF72 does. The other two possibilities revolve around a gain-of-function premise. In one scenario, the RNA foci muck up the expression of a plethora of genes by sequestering and withholding the services of crucial RNA-binding proteins, which play key roles in everything from RNA splicing to transport to translation. In the other, the expansion exerts its toxicity at the protein level: DPRs somehow damage neurons, either through the inclusions they form or, more provocatively, through soluble forms that researchers have yet to detect.
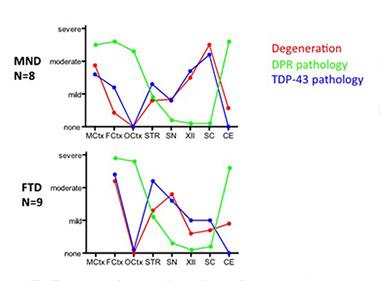
Where Do DPRs Fit In?
When severity is plotted against brain regions, TDP-43 pathology (blue) appears nearly in lockstep with neurodegeneration (red) in people with ALS (top panel) or FTD (bottom). In contrast, dipeptide repeats (green) do not line up with neuronal damage. [Image courtesy of Mackenzie et al., Acta Neuropathologica, 2013.]
Leonard Petrucelli of the Mayo Clinic in Jacksonville, Florida made a case implicating the DPRs. In co-discovering that RAN translation could beget DPRs, Petrucelli’s lab developed a C9-RANT antibody against the poly-GP DPR. The antibody revealed the existence of both cytoplasmic and nuclear inclusions in the brains of people with FTD/ALS who carried the C9ORF72 expansions. Unlike the repeat-laden RNA foci the researchers detected in cells throughout the carriers’ bodies, for the most part the DPR inclusions were only present in neurons in the brain (see Ash et al., 2013). Petrucelli interpreted those initial findings as an indication that the RNA foci do not closely correlate with DPR pathology. He hypothesized that transcript levels of the repeats dictated the development of inclusions.
In collaboration with Matthew Disney’s lab at the Scripps Research Institute in Jupiter, Florida, Petrucelli’s lab has since developed small-molecule inhibitors that bind to the RNA repeats and block the formation of both RNA foci and DPRs. They also developed an antibody assay that could detect GP DPRs in the cerebrospinal fluid (CSF) of C9ORF72 expansion carriers with ALS (see Aug 2014 news story). At the meeting, Petrucelli presented new progress on both fronts. He reported that his lab had developed stable cell lines to facilitate additional drug screening of compounds, a process that has so far quadrupled potency, Petrucelli told the crowd.
He has also expanded his CSF biomarker studies to include FTD patients with C9ORF72 expansions through collaborations with other institutions, including the University of California, San Francisco. His early findings include detection of soluble GP DPRs in the CSF of FTD patients, as well as asymptomatic C9ORF72 expansion carriers. GP concentrations rose in some patients from one year to the next, which could hint that DPRs are associated with progressive pathology.
During the ICFTD, the NIH announced it had given a P01 grant to Petrucelli and his Mayo Clinic colleagues to advance C9FTD/ALS research, which will award the researchers $1.3 million during the first year (see Part 4 of this series).
Adrian Isaacs of University College London reviewed recently published data implicating the DPRs in neurodegeneration in a Drosophila model. He had reported that the DPRs were both necessary and sufficient to cause C9ORF72-mediated neurodegeneration in the fly eye; RNA foci alone were not enough (see Aug 2014 news story).
A fly in this ointment came from Chris Shaw of Kings College London, who presented data that seemed to question the role of C9ORF72 DPR inclusions in neurodegeneration. Shaw first recapped results from a paper last year, in which his group had reported that RNA foci sequestered a number of key RNA-binding proteins and killed neurons in culture, whereas the cells that managed to translate DPRs fared better (see Lee et al., 2013). He also presented some data in which some DPRs, when expressed at high enough levels, could cause toxicity in cell culture.
However, Shaw said the connection between DPR inclusions and disease started to crumble when researchers compared the distribution of inclusions to that of neuropathology in the diseased human brain. To make this point, Shaw drew on dramatic data from a study led by Manuela Neumann of the University of Tübingen, Germany. In that study, which Neumann referenced in her talk at the conference, researchers compared the distribution of C9ORF72 GA DPR inclusions to that of TAR-DNA-binding protein 43 (TDP-43) inclusions and to neurodegeneration in C9ORF72 carriers with FTD, FTD/ALS, or ALS. As had been reported previously, they found that TDP-43 built up in cytoplasmic inclusions in people with these disorders, regardless of whether they harbored C9ORF72 expansions. The TDP-43 inclusions are thought to be toxic to cells.
Importantly, Neumann found that where there were TDP-43 inclusions, there was also neurodegeneration. Both were highest in the motor cortex and spinal cord in ALS patients, and in the frontal cortex in FTD patients. However, the distribution of C9ORF72 GA dipeptide repeat inclusions correlated hardly at all with neurodegeneration. It was similar between patients with FTD or ALS, with levels highest in the cerebellum, frontal and occipital cortices (see Mackenzie et al., 2013). Shaw went on to present new data revealing via western blot that the amount of insoluble TDP-43 inclusions was largely the same in FTD patients with or without C9ORF72 expansions, further disentangling C9ORF72 DPRs (at least GA) from TDP-43 pathology.
Although the role of TDP-43 in neurodegeneration is still not clear, the fact that it seems entwined with neuronal death indicates that it may arise late in the disease process. In support of this idea, Petrucelli mentioned a study led by Mackenzie in which a single C9ORF72 carrier died of a pulmonary embolism at the age of 26. He had childhood learning problems and became psychotic prior to his death, but had not been diagnosed with FTD. At autopsy, his brain was riddled with C9ORF72 GA DPRs, but not yet with TDP-43 inclusions (see Proudfoot et al., 2014). How C9ORF72 expansions are related to TDP-43 pathology—and why that TDP-43 pathology also occurs in patients who do not have C9 expansions—remains a mystery. Some researchers even suggested that the presence of DPRs could be a sign of a protective, rather than destructive, mechanism in certain neurons, for example in the cerebellum.
How should researchers proceed to uncover the role of the C9ORF72 in FTD/ALS? For starters, they would be well advised to compare the distribution of all known DPRs, not just focus on one or another, commented Ian Mackenzie of the University of British Columbia in Vancouver. Mackenzie co-authored the C9 distribution study with Neumann. He said that it will be important to determine whether soluble forms of the DPRs, rather than the more easily visible neuronal inclusions, play a role in neurodegeneration.
RNA foci are more widely distributed than either DPRs or TDP-43 inclusions. Mackenzie added that perhaps a key factor to consider is which cell populations are most susceptible. For example, although RNA foci can be found throughout the body, perhaps only some neuronal populations are sensitive to the sequestration of certain RNA binding proteins, he said. This selective vulnerability could dictate where in the brain neurodegeneration strikes.
Some of these questions might get answers with new mouse models presented at the conference. Clotilde Lagier-Tourenne of the University of California, San Diego, presented initial findings from transgenic mice expressing either 110 or 450 C9ORF72 repeats. She found RNA foci and DPRs only in the mice expressing 450 repeats. Boosting their expression level slightly enhanced RNA foci and DPR expression. Lagier-Tourenne plans to use these mice to study the relationship between C9ORF72 expansions and neurodegeneration, and to test whether targeting the repeats with antisense oligonucleotide (ASO) therapies can prevent it.
ASOs are RNA-like chemicals that are modified at the backbone to make them bind more potently to their target mRNAs. At ICFTD, ASOs got praise as an innovative way forward. Tim Miller of Washington University, St. Louis, presented data showing that targeting normal tau in this way to reduce tau protein levels improved a list of outcome markers in mouse tauopathy models from pathology to neuron loss, astrogliosis, and even seeding. An ASO therapy against SOD1 performed well on safety and pharmacokinetics in a Phase 1 trial for amyotrophic lateral sclerosis (ALS) (see Miller et al., 2013). Plans to develop ASOs against C9ORF72 are only just beginning, said Mackenzie, but the concept makes great sense. With C9ORF72, scientists could target only the pathogenic expansion. “If the expansion exerts a toxic gain of function, which most of us think, then you can prevent that,” said Mackenzie.
Reporting from the mouse front, Adam Walker of the University of Pennsylvania presented striking findings from a new TDP-43 transgenic line. These animals express a doxycycline-controllable TDP-43 gene that lacks its nuclear localization signal, relegating the protein to the cytoplasm. The mice rapidly deposited phospho-TDP-43 inclusions throughout the brain and spinal cord shortly after researchers switched on the gene. Severe motor-neuron disease followed soon, and the mice died by three months of age. The researchers can rescue motor deficits by switching off the toxic gene. Walker said he will use the mice to understand how TDP-43 pathology causes neurodegeneration, and to test out new therapeutics. One can’t help but wonder what might happen if Walker and Lagier-Tourenne’s mice got together.—Jessica Shugart
References
News Citations
- Corrupt Code: DNA Repeats Are Common Cause for ALS and FTD
- RNA Twist: C9ORF72 Intron Expansion Makes Aggregating Protein
- Second Study Sees Intron in FTLD Gene Translated
- Drug and Biomarker Candidates for C9ORF72 ALS and FTD
- Meet the Artful Leftie: NIH Jump-Starts U.S.-Canadian FTLD Cohorts
- C9ORF72’s Dirty Work Done by Problem Proteins
Conference Coverage Series Citations
Alzpedia Citations
Paper Citations
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chiò A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, Morrison KE, Pall H, Talbot K, Ansorge O, , Hernandez DG, Arepalli S, Sabatelli M, Mora G, Corbo M, Giannini F, Calvo A, Englund E, Borghero G, Floris GL, Remes AM, Laaksovirta H, McCluskey L, Trojanowski JQ, Van Deerlin VM, Schellenberg GD, Nalls MA, Drory VE, Lu CS, Yeh TH, Ishiura H, Takahashi Y, Tsuji S, Le Ber I, Brice A, Drepper C, Williams N, Kirby J, Shaw P, Hardy J, Tienari PJ, Heutink P, Morris HR, Pickering-Brown S, Traynor BJ. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012 Apr;11(4):323-30. PubMed.
- Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013 Feb 20;77(4):639-46. PubMed.
- Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C, Adachi Y, Sardone V, Miller JW, Smith BN, Gallo JM, Ule J, Hirth F, Rogelj B, Houart C, Shaw CE. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013 Dec 12;5(5):1178-86. Epub 2013 Nov 27 PubMed.
- Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, Weng SM, Haass C, Kretzschmar HA, Edbauer D, Neumann M. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 2013 Dec;126(6):859-79. Epub 2013 Oct 6 PubMed.
- Proudfoot M, Gutowski NJ, Edbauer D, Hilton DA, Stephens M, Rankin J, Mackenzie IR. Early dipeptide repeat pathology in a frontotemporal dementia kindred with C9ORF72 mutation and intellectual disability. Acta Neuropathol. 2014 Mar;127(3):451-8. Epub 2014 Jan 21 PubMed.
- Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, Andres PL, Mahoney K, Allred P, Alexander K, Ostrow LW, Schoenfeld D, Macklin EA, Norris DA, Manousakis G, Crisp M, Smith R, Bennett CF, Bishop KM, Cudkowicz ME. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013 May;12(5):435-42. Epub 2013 Mar 29 PubMed.
Further Reading
Papers
- Stepto A, Gallo JM, Shaw CE, Hirth F. Modelling C9ORF72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol. 2014 Mar;127(3):377-89. Epub 2013 Dec 24 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.