News on Tau: More Mice Enter the Picture, Structure Takes Shape
Quick Links
The Mandelkow group has been working to generate inducible, TET-off tau-transgenic mice to extend their body of cell-based findings into in-vivo data. At the meeting, Magda Mocanu presented an initial analysis. The group had generated four strains, each expressing endogenous mouse tau plus a modest level (0.5 to 2 times endogenous tau) of a different transgene in their forebrain. The set consists of a full-length and an “abbreviated” version of two types of construct. The abbreviated version involves expression of only the repeat domain of the transgene in question. The transgenes are
- a ΔK280 deletion mutation that aggregates aggressively and causes a clinical tauopathy (Rosso et al., 2003) that is, the “proaggregation” strain; and
- an “antiaggregation” control obtained by replacing two residues in tau’s most aggregation-prone hexapeptide motifs with prolines, which prevents formation of β-structure and aggregation (von Bergen et al., 2001).
The researchers first asked how aggregation influences pathology. In the case of the full-length proaggregation form of tau, the scientists saw a pathological conformation of tau (i.e., MC1 staining) and hyperphosphorylation at many sites in the hippocampus after 6 months, but no tangle formation. They saw only minor changes in phosphorylation with the antiaggregation mutant. All changes were reversible when tau expression was switched off for 6 weeks. In the case of the short form of tau, the proaggregation mutant began developing tangles in the entorhinal cortex at 3 months and progressed with age, even at low levels of exogenous tau (0.7 of endogenous), whereas the antiaggregation mutants showed no tangles. The proaggregation short form of tau nucleated aggregates, which then grew to incorporate both transgenic and endogenous tau. In old mice, the proaggregation, but not the antiaggregation mutants caused synaptic loss and neurodegeneration in the dentate gyrus of the hippocampus. In addition, this tau showed the redistribution from the axonal to the somatodendritic compartment that is characteristic of AD, and it showed enhanced phosphorylation at the KXGS motifs that control microtubule interactions. Switching off the short proaggregation form of tau for 6 weeks after 10 months of expression reversed the mislocalization and phosphorylation of soluble tau, but the aggregated protein remained largely in place. A direct comparison with other tau-transgenic mouse strains, including those made by the Davies, Ashe, and LaFerla labs, can be made once analysis of these mice is more advanced.
Fred Van Leuven of the University of Leuven, Belgium, presented new data in support of his view that tangles may not be the primary offender in tauopathy. Instead, Van Leuven said, the true causes of neurodegeneration—while still elusive—appear prior to, or in parallel to, the tangles. A similar shift is well underway in the amyloid field (Moechars et al., 1999; Dewachter et al., 2002 and many other investigators), but is only beginning in the tau field. For one, Van Leuven presented data on a mouse line that expresses both the tau kinase GSK3β and the human tauopathy mutation P301L. These double-transgenics display a heavy load of neurofibrillary tangles in their hippocampus and cortex, showing them in practically every neuron visible in microscopic sections. Yet despite this massive pathology, the mice survive normally, probably because GSK3β overexpression somehow lessened tauopathy in the mice’s hindbrain and spinal cord. (Mice transgenic for P301L alone do die early, see Terwel et al., 2004; the authors believe that the tauopathy in hindbrain kills them.) Besides emphasizing that regional differences between different brain areas contribute to what ends up being pathogenic, these data suggest that tangles per se do not necessarily cause neurodegeneration, said Van Leuven. Other factors, some having to do with a predeposition interplay between Aβ and tau, do the neurons in, and they still need to be discovered.
Along this line of thought, Van Leuven crossed APP-V717 mice, which have intraneuronal amyloid, diffuse and neuritic plaques, and vascular amyloid, with P301L mice that show intraneuronal tau filaments and die at 1 year of age. Initial analysis of this new, combined model indicates that amyloid pathology is more extensive, and tau pathology dramatically more intense in the forebrain than in the respective single-transgenic lines. Curiously, though, this combined model, too, has less tauopathy in the hindbrain, than the P301L single-transgenics and the doubly afflicted mice live longer, too. Confusing as this is, said Van Leuven, these two studies have in common that they both point to GSK3β as a factor linking Aβ and tau pathogenesis. This connection has been made by studies of lithium and of Aβ oligomer immunization (see, e.g., Ma et al., 2006).
Finally, Van Leuven added new data to the study of p25/Cdk5. Prior work by Li-Huei Tsai, Van Leuven, and others has put this kinase on the AD map, and LaFerla’s group has linked fibrillar Aβ to inflammation and tau hyperphosphorylation via p25/Cdk5 (Kitazawa et al., 2005). At the Eibsee, Van Leuven added data by showing that inducible p25 transgenic mice suffer severe neurodegeneration with drastic loss of brain volume that is mediated not primarily by tauopathy but by early microgliosis and astrogliosis. In yet another demonstration that transgenic mice can be unpredictable and bewildering, Tsai’s inducible p25 transgenic mice do show tau pathology (Cruz et al., 2003). Rather than being a model for tauopathy, Van Leuven views his new mouse as a window into hippocampal sclerosis, a common cause of adult epilepsy (see Sen et al., 2006).
Frank LaFerla reviewed recently published research on his group’s ongoing analysis of the triple-transgenic mouse model (e.g., Oddo et al, 2006; Billings et al., 2005). He emphasized that, overall, this study to date suggests that Aβ facilitates subsequent tau pathology by three pathways.
- One, fibrillar Aβ tends to act via inflammatory processes in that amyloid induces cytokines that, in turn, activate the tau kinase Cdk5 (Kitazawa et al., 2005).
- Second, LaFerla believes that Aβ oligomers interfere with the proteasome’s ability to clear tau (Oddo et al., 2004). On this issue, Haass questioned how intraneuronal Aβ would interact with the proteasome from its primary site of residence within the lumen of endosomes and late endosomes (also called multivesicular bodies). LaFerla noted that the triple-transgenic mice have large quantities of Aβ across different intraneuronal organelles, even some in the cytosol. A study published last month by Gunnar Gouras’s lab may shed light on this issue (see Almeida et al., 2006). In neurons from human AD brain, Aβ was spotted in multivesicular bodies (Takahashi et al., 2002).
- Third, various tau kinases, including GSK3β, become more active in response to rising Aβ levels (Oddo et al., 2006).
- Theoretically, axonal transport effects might constitute a fourth link, but data on that question is not available yet.
Importantly, the phosphorylation state of tau, more than its absolute levels, determines whether tau pathology can reverse after Aβ removal. Stimulation of the cholinergic system with M1 agonists also affects both pathologies (Caccamo et al., 2006). This approach will see its definitive test in the clinic as TorreyPines Pharmaceuticals tests NXG267; other pharmaceutical companies are said to take a second look at such agonists, as well.
Clearly, compensatory processes wrestle with toxic ones at the early stages of tau pathology, and their outcome determines the cell’s fate. One path toward understanding how tau goes bad early on is to understand its structure, a line of inquiry spearheaded by Eckhard Mandelkow. At the conference, Mandelkow started by noting that the repeat domain of tau is central to its aggregation. Imaging tau is notoriously difficult because tau is natively unfolded. It is highly soluble, and probably resists folding because it contains unusually few hydrophobic amino acids for a protein of its size. Indeed, its partially aggregated oligomeric forms clearly come up in Mocanu’s transgenic mice prior to paired helical filaments, but even they are unstable and too floppy to be viewed in the EM, as has been done for other amyloid oligomers, let alone be crystallized for x-ray imaging.
How to nail this pudding to the wall, then? The Mandelkow group has used other structural approaches to get a feeling for tau’s conformations. Tau contains two hexapeptide motifs flanking the repeat 2 domain that form stable β structures and seed tau aggregation. The ΔK280, as well as the P301L mutation responsible for FTDP-17, make precisely those areas more hydrophobic and prone to assume a β-structure (von Bergen et al, 2001; von Bergen et al., 2005). Mandelkow calls these motifs VQIINK and VQIVYK (pronounced VIKING and VIKIVIK, respectively). Their study received an independent boost last year when David Eisenberg at the University of California, Los Angeles, published their crystal structure (Nelson et al., 2005). It was taken from a prion protein and the paper related primarily to the structural conversion of cellular prion protein to its pathogenic alter ego, yet a similar motif occurs in Aβ and in tau (see also Inouye et al., 2006). Eisenberg proposed a broader concept of an “amyloid spine,” whereby in all these amyloid building blocks the hydrophobic side chains of the seeding motif face each other to form a tight surface that interacts through van der Waals forces. Called a steric zipper, it is based in part on the principle of protein folding that hydrophobic residues tend to turn inside, in this case in the form of two β strands leaning against each other. Eisenberg’s framework fits with Mandelkow’s own model of these kernels of nascent β-structure, as well as with his group’s simultaneous description of the tau repeat domain by NMR solution structure (Mukrasch et al., 2005, done in collaboration with Christian Griesinger’s group at the Max-Planck Institute for Biophysical Chemistry in Goettingen).
Another way of making educated guesses about a protein’s structure is FRET, a method where scientists attach pairs of fluorophores along a protein’s length and then measure fluorescence when these pairs come close together as the protein folds. With this approach, Mandelkow’s group recently found that tau in solution does not actually exist as a totally random coil as one might expect for natively unfolded proteins. Instead, it assumes a sort of paper clip structure that, while still flexible, nevertheless shows the C-terminus bending over to lie in the middle between the repeat domain and the N-terminus, close enough to give FRET with both (Jeganathan et al., 2006; see image below). 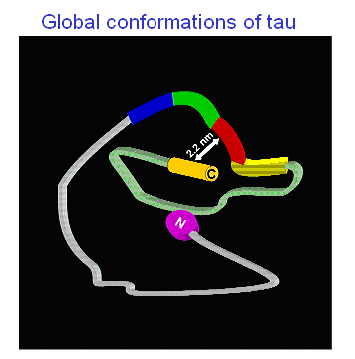
It’s still unclear how this paper clip structure relates to early AD pathology, but the widely used tau antibodies MC1 and Alz50 (Jicha et al., 1997) provide an intriguing clue, Mandelkow said. They are thought to pick up a misfolded and pathogenic, but pretangle, conformation of tau; they, too, recognize a composite epitope made up of amino acids from both the N-terminal and the repeat domain of tau.—Gabrielle Strobel.
References
Therapeutics Citations
Paper Citations
- Rosso SM, Donker Kaat L, Baks T, Joosse M, de Koning I, Pijnenburg Y, de Jong D, Dooijes D, Kamphorst W, Ravid R, Niermeijer MF, Verheij F, Kremer HP, Scheltens P, van Duijn CM, Heutink P, van Swieten JC. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003 Sep;126(Pt 9):2016-22. Epub 2003 Jul 22 PubMed.
- von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow EM, Mandelkow E. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J Biol Chem. 2001 Dec 21;276(51):48165-74. PubMed.
- Moechars D, Dewachter I, Lorent K, Reversé D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999 Mar 5;274(10):6483-92. PubMed.
- Dewachter I, Reversé D, Caluwaerts N, Ris L, Kuipéri C, Van den Haute C, Spittaels K, Umans L, Serneels L, Thiry E, Moechars D, Mercken M, Godaux E, Van Leuven F. Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J Neurosci. 2002 May 1;22(9):3445-53. PubMed.
- Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, Van Leuven F. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J Biol Chem. 2005 Feb 4;280(5):3963-73. PubMed.
- Ma QL, Lim GP, Harris-White ME, Yang F, Ambegaokar SS, Ubeda OJ, Glabe CG, Teter B, Frautschy SA, Cole GM. Antibodies against beta-amyloid reduce Abeta oligomers, glycogen synthase kinase-3beta activation and tau phosphorylation in vivo and in vitro. J Neurosci Res. 2006 Feb 15;83(3):374-84. PubMed.
- Kitazawa M, Oddo S, Yamasaki TR, Green KN, Laferla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer's disease. J Neurosci. 2005 Sep 28;25(39):8843-53. PubMed.
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003 Oct 30;40(3):471-83. PubMed.
- Oddo S, Caccamo A, Smith IF, Green KN, Laferla FM. A dynamic relationship between intracellular and extracellular pools of Abeta. Am J Pathol. 2006 Jan;168(1):184-94. PubMed.
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005 Mar 3;45(5):675-88. PubMed.
- Oddo S, Billings L, Kesslak JP, Cribbs DH, Laferla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004 Aug 5;43(3):321-32. PubMed.
- Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006 Apr 19;26(16):4277-88. PubMed.
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002 Nov;161(5):1869-79. PubMed.
- Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, Laferla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem. 2006 Jan 20;281(3):1599-604. PubMed.
- Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, Laferla FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006 Mar 2;49(5):671-82. PubMed.
- von Bergen M, Barghorn S, Biernat J, Mandelkow EM, Mandelkow E. Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim Biophys Acta. 2005 Jan 3;1739(2-3):158-66. PubMed.
- Nelson R, Sawaya MR, Balbirnie M, Madsen AØ, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005 Jun 9;435(7043):773-8. PubMed.
- Inouye H, Sharma D, Goux WJ, Kirschner DA. Structure of core domain of fibril-forming PHF/Tau fragments. Biophys J. 2006 Mar 1;90(5):1774-89. PubMed.
- Mukrasch MD, Biernat J, von Bergen M, Griesinger C, Mandelkow E, Zweckstetter M. Sites of tau important for aggregation populate {beta}-structure and bind to microtubules and polyanions. J Biol Chem. 2005 Jul 1;280(26):24978-86. PubMed.
- Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006 Feb 21;45(7):2283-93. PubMed.
- Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997 Apr 15;48(2):128-32. PubMed.
Further Reading
News
- Tall Science at Small Retreat: Dispatch from Germany’s Eibsee
- Oligomers Abound, But Where’s the Seed?
- γ-Secretase: Ins and Outs of a Voracious Membrane Protein Grinder
- First Look at the Secretase, New Kid on the Block
- Assembly, Traffic Escorts, Fats All Control APP Processing
- An Intricate Dance: α-Secretase and Its Partners
- Just How Does APP Build a Better Brain?
- Have APP, Will Travel
- Shedding Assumes α Status
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.