Detecting Familial AD Ever Earlier: Subtle Memory Signs 15 Years Before
Quick Links
Recent findings have strengthened the scientific underpinning of the Alzheimer’s Prevention Initiative (API). On 3 February 2011, Natalia Acosta-Baena and colleagues reported in the Lancet Neurology that they were able to capture a clear decline in cognition starting in people’s early thirties in the largest-known population with autosomal-dominant Alzheimer’s disease. They define an earlier disease stage prior to what is called pre-MCI, in effect pushing the line of detectability back toward younger ages by some four years. Two other papers go in the same direction. Last July in the journal Brain, Mario Parra and colleagues published a new test that appears to detect a specific visual memory deficit perhaps even earlier, at ages when mutation carriers perform as well as controls on standard neuropsychometric tests. And in last December’s Annals of Neurology, Yakeel Quiroz and colleagues report the first of what is expected to be a wave of preclinical brain imaging findings. Carriers in their thirties, while still performing the memory test at hand as well as non-carriers, drive their hippocampus harder to achieve that parity. Together, these three papers push back the preclinical phase of AD that is amenable to detection by way of neuropsychology and imaging. They characterize the 20 to 15 years prior to dementia in greater detail. All three are coauthored by Francisco Lopera, a neurologist at Universidad de Antioquia, Medellin, Colombia, who has characterized and cared for the largest known population in the world with autosomal-dominant AD (ADAD) inherited via a presenilin-1 mutation.
Being based in South America, Lopera is little known among researchers elsewhere, but in fact, his group has published quality research in highly regarded journals for decades. For example, just last December Lopera published results of a collaboration with Markus Glatzel at University Medical Center in Hamburg, Germany, and researchers at Novartis in Basel, Switzerland, and the University of Barcelona, Spain, which found that the cerebellum is more affected in AD than previously thought, raising questions about its widespread use as a reference region; see Sepulveda-Falla et al., 2010.
The Acosta-Baena, Parra, and Quiroz papers reflect different facets of the field’s combined push toward refining the still-sketchy description of the pre-dementia phase of AD (Acosta-Baena et al., 2011; Parra et al., 2010; Quiroz et al., 2010). This is necessary to upgrade clinicians’ toolkits for measuring whether an experimental drug does any good in future pre-symptomatic trials. Toward that goal, Jessica Langbaum of the Banner Alzheimer’s Institute in Phoenix, Arizona, reported a fourth sign of progress at a 7 January 2011 conference of the Alzheimer’s Prevention Initiative held in Washington, D.C. Together with colleagues at Banner and biostatistician Suzanne Hendrix of Pentara Corporation in Salt Lake City, Utah, Langbaum and the team built upon Hendrix’s previous findings that the trusty but imperfect ADAS-Cog package of neuropsychometric tests performs better in clinical trials of MCI and AD if the scientists remove certain components that add variability and dilute the battery’s predictive power (Hendrix and Wells, ICAD 2010). With these and other efforts, scientists should soon be able to put together a serviceable package of outcome measures for secondary prevention trials, Lopera said. Read on for a summary of the three papers and Langbaum’s study.
Fifteen Years of Observation on 449 Carriers
Many scientific groups these days are trying to define the symptoms and biomarker changes in the years before dementia. “The diagnosis of AD is marching leftward on the time scale,” said Paul Aisen of the University of California, San Diego. Different groups take different approaches, and, consequently, parallel terminologies coexist in the field to denote otherwise similar stages of pre-dementia (Dubois et al., 2010; ARF related ICAD story). In the Lancet Neurology paper, Lopera and his team framed their study in the language of mild cognitive impairment (MCI). Because it is clear that some cognitive deficits precede MCI as defined by its diagnostic criteria, the stage of pre-MCI has been suggested (Reisberg et al., 2008). In the general population, the MCI category is heterogeneous because a significant fraction of patients do not progress to AD or even revert from MCI back to normal. In contrast, all previous longitudinal studies of familial AD have noted a continuous, gradual decline, making inherited AD an ideal model to characterize the long slide into dementia from its very beginning.
Each of the previous familial AD studies was small. In this paper, the Colombian scientists retrospectively analyzed descendents of the largest-known cohort of autosomal-dominant AD, including 1,784 patients age 17 to 70 who came to Lopera for treatment and research between 1995 and 2010. This study is by far the biggest study of its kind. Four hundred forty-nine people carried the E280A Paisa mutation. Four hundred ninety-nine non-carriers served to establish normal parameters on the expanded CERAD battery of cognitive tests that the scientists administered to the participants at follow-ups every other year where possible. The scientists modified and added some tests to the U.S.-based CERAD battery to adapt it to the language and educational differences of this Colombian population. These people have from one to 11 years of education and low income, though as a group they function well in life.
What were the very first signs the researchers saw? Memory loss in the early thirties, Lopera said. To describe this finding, the scientists ended up splitting the published pre-MCI category into two distinct stages. “We were originally looking only for MCI and pre-MCI, but when we analyzed the data, we found that there are people who are otherwise asymptomatic but in one test fall short by two standard deviations. They do not yet meet criteria for pre-MCI,” Lopera told this reporter. These are people who do not complain of memory problems yet and have no functional impairment, but they do fail one objective measure in the CERAD-plus battery. Seventeen people fell into this group in this analysis. Anecdotally, Dominantly Inherited Alzheimer Network (DIAN) investigators have noted that in their hands, too, some otherwise asymptomatic participants send up an early flag by doing poorly on a single test. Longitudinal research on both the DIAN and Paisa cohort is ongoing.
How did the Paisa mutation carriers progress from that first cognitive deficit? Some four years later, around age 38, they tended to meet published criteria for pre-MCI; six years later, at age 44, they had progressed to MCI; and five years later still, around 49, to dementia. They then live with dementia for an average of 10 years. The very first deficits can show up in a variety of memory domains.
Overall, the cognitive profile of this population is very similar to that of sporadic AD, Lopera said. This means that about 25 years pass between this first (at present) measureable sign of a cognitive problem and death from AD. The age at each stage varies somewhat from person to person, but the range is relatively small, around three to four years.
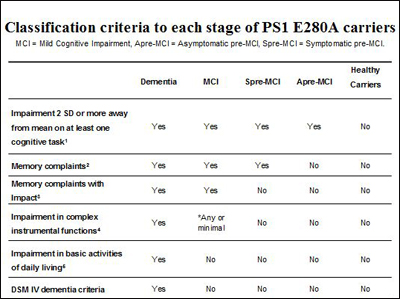
Presented at a 7 January 2011 API conference in Washington, D.C., this slide recapitulates the stages of autosomal-dominant AD in the Colombian carriers of the presenilin Paisa mutation, formally published on 3 February 2011. Image credit: Francisco Lopera
“The systematic description of the natural disease history in this cohort provides a framework for the design of studies of preventive intervention,” writes John Ringman of the University of California, Los Angeles, in a comment in the same issue of Lancet Neurology. Ringman leads a DIAN site and has studied different presenilin-1 families in Southern California and Mexico with cognition and biomarker measurements (e.g., Murrell et al., 2006).
A New Test
The Acosta-Baena study pushes back the time of the first detectable sign of AD compared to a previous, smaller analysis of people with the Paisa mutation, which had pegged subjective memory complaint to around age 38 (Ardila et al., 2000). But it is not the last word on the matter. The protocol used in this study was designed for dementia; it is less sensitive in the preclinical years than newer tools scientists across the field are developing. One such tool comes from Lopera’s group itself. Probing a fundamental function of visual short-term memory that is not part of any standard psychometric battery, the new test shows a robust effect already during a carrier’s early thirties, the age at which they also show the first decrement in a conventional memory test that Acosta-Baena et al. report.
Developed by Mario Parra, then a student in Lopera’s group, this new tool tests a person’s visual short-term memory binding. That is a memory function that allows people to “bind” together moment-to-moment changes in shapes and colors in their memory temporarily, for example, to remember whether they just took the yellow or the white pill. It is different from remembering stable properties of the world, for example, the shape and color of your mailbox or of the school bus. Previous research had shown that short-term visual memory binding is independent of aging. This seemed an advantage over other associative learning tests that are widely used in AD research, because these do decline with normal aging and hence make it more difficult to pin a change firmly on preclinical AD.
To test whether visual short-term memory binding flags future AD early, Parra enrolled 22 symptomatic Paisa mutation carriers, 30 asymptomatic carriers in their thirties, and 30 matched non-carriers. He subjected them both to a fleet of nine established memory tests and to a binding test in which they learned and then recalled both the shape and the color of polygons on a computer screen. Asymptomatic carriers were not statistically different from the non-carriers on any of the standard neuropsychological tests (though they trended downward a little), but they did have striking trouble with the binding tests. They were able to recall shapes all right; they were able to recall colors all right, but they could not bind the two together in their memory. The non-carriers could. In the binding task, the asymptomatic carriers performed just like symptomatic carriers. Among these 10 tests, then, only short-term visual binding distinguished the young asymptomatic carriers from non-carriers, and it did so clearly, the scientists report.
The test is surprisingly sensitive and specific for AD, Lopera said. Unlike other neuropsychometric tests, it does not vary with socioeconomic background or education. In its published form, the test is not practical because it takes some 80 minutes to administer. But since then, Parra has developed a 15-minute version, which in unpublished tests of 40 carriers and 80 non-carriers works as well, Lopera said. Like any new finding, this will have to be reproduced independently in other cohorts before scientists across the field accept it. Lopera hopes that will happen. Previous research indicates that visual short-term binding may well detect sporadic Alzheimer’s at the pre-MCI stage, too. Ongoing testing in the Colombian families will show whether this test distinguishes carriers from non-carriers even earlier than the two standard deviation decline reported as the first sign by Acosta-Baena et al.
There is a desire across the field to agree on tests that are sensitive and robust in the earliest stages of the AD process. Regulators keep saying, and scientists recognize, that cognitive tests will be needed to bridge the gap between expected drug effects on preclinical biomarkers and the eventual clinical benefit that matters to the patient and that allows a new treatment to be approved. It is not realistic to show that a drug hits a target in the desired way in a wholly asymptomatic person, and then wait many years to see if the clinical diagnosis changes. Change in early cognitive tests should accompany biomarker outcomes, or follow on within a practical time period. Between these recent papers and other research, Lopera sees a combined episodic memory and executive phenotype that should yield suitable tests soon. (Visual short-term binding is an episodic memory function.)
Imaging Markers in Paisa Mutation Pedigrees
What about biomarkers in these thirty-something carriers, anyway? The API researchers are gearing up for an ongoing study in which they intend to characterize the major markers in the Colombian population across all age ranges, first cross-sectionally and then longitudinally going forward. They aim to pinpoint for each marker at what age carriers first diverge from non-carriers, and to track the changes from there to dementia. To date, one brain imaging paper in the December 2010 Annals of Neurology and a Society for Neuroscience 2010 conference abstract are published. In the paper, Yakeel Quiroz, a Ph.D. student and collaborator of Lopera’s who is now at Boston University in the laboratory of Chantal Stern, reports that young carriers of the Paisa mutation activate their right anterior hippocampus more strongly than age-matched non-carriers as they encode face-name associations in an established functional MRI task (Quiroz et al., 2010). The SfN abstract (Quiroz et al., 2010) expands the finding to include the right dorsolateral prefrontal cortex and the left lateral parietal cortex. These carriers were in their thirties, with a mean age of 33.7. They perform as well as the non-carriers, but, in essence, for that their brain has to work harder. The volume of their hippocampus at that age did not differ from that of controls.
This study substantiates an earlier paper by the Swiss scientist Katrin Henke and colleagues, who had also found hippocampal hyperactivation in young pre-symptomatic carriers of a different presenilin-1 AD mutation. Henke’s study, like most prior research on individual families, was small, including but two probands (Mondadori et al., 2006). Quiroz’s paper reports on 20 carriers and 19 non-carrier family controls, bulking up the data considerably. Researchers led by Ringman also recently published fMRI data in Mexican families with a different presenilin-1 mutation. They found activation going up as carriers approached their expected age at onset (Braskie et al., 2010). Most other familial AD imaging studies are small, but that makes them no less interesting. Just last month, Swedish researchers reported that glucose metabolism went down over time in two carriers of a different presenilin-1 mutation. This happened in the posterior cingulate, the parietal, and parietotemporal cortex, and accompanied subtle cognitive decrements; see Schöll et al., 2011.
Quiroz’s results in the Colombian families speak to a debate among researchers about how brain activation changes in the decade or more before AD’s dementia phase. Findings particularly at the MCI stage have shown variable results, perhaps because change happens in biphasic curves over time, or because different regions change at different times, or because the patients in those studies were heterogeneous. More recent fMRI studies have coalesced around the notion that an early phase of hippocampal hyperactivation precedes a later hypoactivation, a biphasic curve that, to the popular imagination, suggests a brain that struggles mightily to keep up before it crashes (e.g., Dickerson et al., 2005). MCI studies support that in the sense that early stage, mildly impaired people tend to show hyperactivation, whereas more severely impaired MCI patients show hypoactivation (Celone et al., 2006). Autosomal-dominant families can help clarify this issue because they are less heterogeneous and tend to yield more consistent results.
Taken together, this means that by Paisa mutation carriers’ early thirties, some 15 years before they meet the traditional dementia diagnosis, scientists at present have three types of test in hand that distinguish carriers from non-carriers. They are the deficit on a single established cognitive test as reported in Lancet Neurology, the new binding test reported in Brain, and the functional MRI measure reported in Annals of Neurology. All these changes can be measured at what Lopera calls the “asymptomatic pre-MCI” stage, suggesting that it makes sense to start examining individuals at high risk for AD as early as 20 years before the clinical start of the disease.
Ongoing research is doing exactly that, and more. Quiroz is already imaging the brains of children because she saw some signals in teenagers, and fMRI is not invasive. API researchers and collaborators are currently analyzing CSF samples drawn from young adults in their thirties and twenties. For each marker, the scientists compare carriers and non-carries in a given age range and, if they see a difference, push back to the next-younger age range, Lopera said. Collectively, their ambition is to capture the natural trajectory of the disease in its entirety. “During this year, we plan to obtain all these biomarkers in carriers and non-carriers in all the age ranges,” Eric Reiman of the Banner Alzheimer’s Institute in Phoenix, Arizona, said at the API conference in Washington on 7 January 2011.
Don’t Count Out Old Faithful
A geyser it’s not, but the ADAS-Cog has been a trusty instrument to measure cognition in trial after trial of Alzheimer’s disease since the long-gone days of success with the cholinesterase inhibitors. Scientists especially in industry tend to use tools the FDA knows and has found adequate for drug approval before, but they agree that the ADAS-Cog is too crude to pick out those subtlest of changes by which an insidious disease like Alzheimer’s creeps up on a person over the course of many years. Likewise, the CERAD is used widely to gather standardized cognitive information harmonized through the National Alzheimer’s Coordinating Center, but it, also, is considered too insensitive to grasp the slippage from normal to mildly impaired cognition.
Or is it? Perhaps these instruments could be tuned to this purpose? This, in essence, is what Langbaum of the Banner and her colleagues did as part of their prep work for API trials. At the first API advisory meeting in October 2009, Langbaum had shown estimates of statistical power and group sizes for treatment trials that were based on data from the Banner scientists’ own research cohorts. She got advice to use more and larger cohorts and a greater range of measures, and did exactly that. “We wanted to identify which combination of cognitive assessments would most sensitively detect a person’s trajectory of change prior to an AD diagnosis,” Langbaum said. Then she would use that combo to calculate power estimates for preclinical trials.
To do that, Langbaum teamed up with Hendrix, an independent biostatistician. Hendrix had analyzed ADAS-Cog results in MCI and AD drug trials with an approach she developed, called mean-to-standard deviation ratio (MSDR). She found that some components of the ADAS-Cog actually diluted the overall result in early stage cohorts, largely because they varied greatly from person to person but changed little over time. The researchers removed these “noisy” tests and kept only the ones that were most similar among people and changed the most from year to year. That cut the needed sample size for a given trial in half. “In this case, less is more,” Langbaum said.
The researchers, notably Napatkamon (Yui) Ayutyanont, a statistician at Banner, then applied this MSDR analysis to every single test given to members of the 14-year Antioquia cohort that formed the basis of Acosta-Baena et al., 2011. She also applied it to assessments included in the cohort studies at the Rush Alzheimer’s Disease Center of Chicago’s Rush University led by David Bennett. These large studies do not use identical batteries; for example, the Antioquia study uses a modified CERAD, but Langbaum said many individual tests within those batteries are very similar or target the same cognitive domains. The scientists focused the MSDR analysis on people who changed from cognitively normal to mildly impaired, as well as comparing carriers to non-carriers (PS1, E4). In this way, they identified an optimal combination of five tests for this transition, which they will formally present at ICAD this July. The combination was similar for the Antioquia and the Rush cohorts, i.e., for a genetic and a sporadic cohort.
“This set of tests gives us an opportunity to detect cognitive decline using smaller sample sizes than we previously thought,” Langbaum said. The set provides better power to detect a treatment effect within two to three years, Reiman added.
Finally, also at the D.C. meeting, Paul Aisen of the University of California, San Diego, noted work on two further cohorts of normal elders who also can be distinguished cognitively as being on the way toward AD. For one, the 200 controls who enrolled in ADNI turned out to contain some 40 percent who had brain amyloid, and this subgroup not only had more brain atrophy over the period of observation, but they also declined slightly even on the non-optimized cognitive instruments in ADNI. For another, a separate normal aging cohort studied by David Salmon at UCSD also spotted this effect. Salmon had no amyloid CSF or PET data, but he did know the ApoE genotype of the study participants. Taking ApoE4 as a proxy for brain amyloid, he, too, found that the E4-carriers declined on delayed recall and some other tests. “This supports the idea that we can use a cognitive outcome measure in a secondary prevention study in cognitively normal individuals,” Aisen said.
Industry scientists called it a real advance for very early stage trials to have subtle cognitive markers. Viewed in isolation, such subtle cognitive changes would be considered clinically meaningless; however, the 7 January 2011 meeting of the API group featured strong consensus that in the context of all that is known about preclinical AD, these early cognitive markers will help bridge the gap between an initial biomarker response to a therapy and an eventual global outcome in patients who are years away from even an MCI or prodromal AD diagnosis.
Together, research on these tests, old and new, serves as groundwork for drug trials in this population. These, in turn, may help the design of future preclinical treatment trials in sporadic AD if indeed these markers can be used to identify people with the same cognitive and biomarker changes. The families, Lopera, and all API scientists hope that the first such trials will get underway in 2012. For more on API progress, read Part 4 of this series.—Gabrielle Strobel
This is Part 3 of a six-part series. See also Part 1, Part 2, Part 4, Part 5, Part 6. View a PDF of the entire series.
References
News Citations
- Noisy Response Greets Revised Diagnostic Criteria for AD
- Scientists and Regulators Discuss Preclinical AD Trials
- Colombians Come to Fore in Alzheimer’s Research, Mass Media
- A Neurologist’s Devotion Puts Familial AD Research Onto New Plane
- Can Adaptive Trials Ride to the Rescue?
- Time to Open the Kimono—Which Drugs in Preclinical Trials?
Paper Citations
- Sepulveda-Falla D, Matschke J, Bernreuther C, Hagel C, Puig B, Villegas A, Garcia G, Zea J, Gomez-Mancilla B, Ferrer I, Lopera F, Glatzel M. Deposition of hyperphosphorylated tau in cerebellum of PS1 E280A Alzheimer's disease. Brain Pathol. 2011 Jul;21(4):452-63. PubMed.
- Acosta-Baena N, Sepulveda-Falla D, Lopera-Gómez CM, Jaramillo-Elorza MC, Moreno S, Aguirre-Acevedo DC, Saldarriaga A, Lopera F. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer's disease: a retrospective cohort study. Lancet Neurol. 2011 Mar;10(3):213-20. PubMed.
- Parra MA, Abrahams S, Logie RH, Méndez LG, Lopera F, Della Sala S. Visual short-term memory binding deficits in familial Alzheimer's disease. Brain. 2010 Sep;133(9):2702-13. PubMed.
- Quiroz YT, Budson AE, Celone K, Ruiz A, Newmark R, Castrillón G, Lopera F, Stern CE. Hippocampal hyperactivation in presymptomatic familial Alzheimer's disease. Ann Neurol. 2010 Dec;68(6):865-75. PubMed.
- Dubois B, Feldman HH, Jacova C, Cummings JL, Dekosky ST, Barberger-Gateau P, Delacourte A, Frisoni G, Fox NC, Galasko D, Gauthier S, Hampel H, Jicha GA, Meguro K, O'brien J, Pasquier F, Robert P, Rossor M, Salloway S, Sarazin M, de Souza LC, Stern Y, Visser PJ, Scheltens P. Revising the definition of Alzheimer's disease: a new lexicon. Lancet Neurol. 2010 Nov;9(11):1118-27. PubMed.
- Reisberg B, Prichep L, Mosconi L, John ER, Glodzik-Sobanska L, Boksay I, Monteiro I, Torossian C, Vedvyas A, Ashraf N, Jamil IA, de Leon MJ. The pre-mild cognitive impairment, subjective cognitive impairment stage of Alzheimer's disease. Alzheimers Dement. 2008 Jan;4(1 Suppl 1):S98-S108. PubMed.
- Murrell J, Ghetti B, Cochran E, Macias-Islas MA, Medina L, Varpetian A, Cummings JL, Mendez MF, Kawas C, Chui H, Ringman JM. The A431E mutation in PSEN1 causing familial Alzheimer's disease originating in Jalisco State, Mexico: an additional fifteen families. Neurogenetics. 2006 Nov;7(4):277-9. Epub 2006 Aug 5 PubMed.
- Ardila A, Lopera F, Rosselli M, Moreno S, Madrigal L, Arango-Lasprilla JC, Arcos M, Murcia C, Arango-Viana JC, Ossa J, Goate A, Kosik KS. Neuropsychological profile of a large kindred with familial Alzheimer's disease caused by the E280A single presenilin-1 mutation. Arch Clin Neuropsychol. 2000 Aug;15(6):515-28. PubMed.
- Mondadori CR, Buchmann A, Mustovic H, Schmidt CF, Boesiger P, Nitsch RM, Hock C, Streffer J, Henke K. Enhanced brain activity may precede the diagnosis of Alzheimer's disease by 30 years. Brain. 2006 Nov;129(Pt 11):2908-22. PubMed.
- Braskie MN, Medina LD, Rodriguez-Agudelo Y, Geschwind DH, Macias-Islas MA, Cummings JL, Bookheimer SY, Ringman JM. Increased fMRI signal with age in familial Alzheimer's disease mutation carriers. Neurobiol Aging. 2012 Feb;33(2):424.e11-21. PubMed.
- Schöll M, Almkvist O, Bogdanovic N, Wall A, Långström B, Viitanen M, Nordberg A. Time course of glucose metabolism in relation to cognitive performance and postmortem neuropathology in Met146Val PSEN1 mutation carriers. J Alzheimers Dis. 2011;24(3):495-506. PubMed.
- Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, Bertram L, Mullin K, Tanzi RE, Blacker D, Albert MS, Sperling RA. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005 Aug 9;65(3):404-11. PubMed.
- Celone KA, Calhoun VD, Dickerson BC, Atri A, Chua EF, Miller SL, DePeau K, Rentz DM, Selkoe DJ, Blacker D, Albert MS, Sperling RA. Alterations in memory networks in mild cognitive impairment and Alzheimer's disease: an independent component analysis. J Neurosci. 2006 Oct 4;26(40):10222-31. PubMed.
Other Citations
External Citations
Further Reading
Papers
- Sepulveda-Falla D, Matschke J, Bernreuther C, Hagel C, Puig B, Villegas A, Garcia G, Zea J, Gomez-Mancilla B, Ferrer I, Lopera F, Glatzel M. Deposition of hyperphosphorylated tau in cerebellum of PS1 E280A Alzheimer's disease. Brain Pathol. 2011 Jul;21(4):452-63. PubMed.
- Pitchumoni SS, Doraiswamy PM. Current status of antioxidant therapy for Alzheimer's Disease. J Am Geriatr Soc. 1998 Dec;46(12):1566-72. PubMed.
- Parra MA, Abrahams S, Logie RH, Méndez LG, Lopera F, Della Sala S. Visual short-term memory binding deficits in familial Alzheimer's disease. Brain. 2010 Sep;133(9):2702-13. PubMed.
- Quiroz YT, Budson AE, Celone K, Ruiz A, Newmark R, Castrillón G, Lopera F, Stern CE. Hippocampal hyperactivation in presymptomatic familial Alzheimer's disease. Ann Neurol. 2010 Dec;68(6):865-75. PubMed.
- Dubois B, Feldman HH, Jacova C, Cummings JL, Dekosky ST, Barberger-Gateau P, Delacourte A, Frisoni G, Fox NC, Galasko D, Gauthier S, Hampel H, Jicha GA, Meguro K, O'brien J, Pasquier F, Robert P, Rossor M, Salloway S, Sarazin M, de Souza LC, Stern Y, Visser PJ, Scheltens P. Revising the definition of Alzheimer's disease: a new lexicon. Lancet Neurol. 2010 Nov;9(11):1118-27. PubMed.
- Reisberg B, Prichep L, Mosconi L, John ER, Glodzik-Sobanska L, Boksay I, Monteiro I, Torossian C, Vedvyas A, Ashraf N, Jamil IA, de Leon MJ. The pre-mild cognitive impairment, subjective cognitive impairment stage of Alzheimer's disease. Alzheimers Dement. 2008 Jan;4(1 Suppl 1):S98-S108. PubMed.
- Murrell J, Ghetti B, Cochran E, Macias-Islas MA, Medina L, Varpetian A, Cummings JL, Mendez MF, Kawas C, Chui H, Ringman JM. The A431E mutation in PSEN1 causing familial Alzheimer's disease originating in Jalisco State, Mexico: an additional fifteen families. Neurogenetics. 2006 Nov;7(4):277-9. Epub 2006 Aug 5 PubMed.
- Ardila A, Lopera F, Rosselli M, Moreno S, Madrigal L, Arango-Lasprilla JC, Arcos M, Murcia C, Arango-Viana JC, Ossa J, Goate A, Kosik KS. Neuropsychological profile of a large kindred with familial Alzheimer's disease caused by the E280A single presenilin-1 mutation. Arch Clin Neuropsychol. 2000 Aug;15(6):515-28. PubMed.
- Mondadori CR, Buchmann A, Mustovic H, Schmidt CF, Boesiger P, Nitsch RM, Hock C, Streffer J, Henke K. Enhanced brain activity may precede the diagnosis of Alzheimer's disease by 30 years. Brain. 2006 Nov;129(Pt 11):2908-22. PubMed.
- Braskie MN, Medina LD, Rodriguez-Agudelo Y, Geschwind DH, Macias-Islas MA, Cummings JL, Bookheimer SY, Ringman JM. Increased fMRI signal with age in familial Alzheimer's disease mutation carriers. Neurobiol Aging. 2012 Feb;33(2):424.e11-21. PubMed.
- Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, Bertram L, Mullin K, Tanzi RE, Blacker D, Albert MS, Sperling RA. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005 Aug 9;65(3):404-11. PubMed.
- Schöll M, Almkvist O, Bogdanovic N, Wall A, Långström B, Viitanen M, Nordberg A. Time course of glucose metabolism in relation to cognitive performance and postmortem neuropathology in Met146Val PSEN1 mutation carriers. J Alzheimers Dis. 2011;24(3):495-506. PubMed.
- Celone KA, Calhoun VD, Dickerson BC, Atri A, Chua EF, Miller SL, DePeau K, Rentz DM, Selkoe DJ, Blacker D, Albert MS, Sperling RA. Alterations in memory networks in mild cognitive impairment and Alzheimer's disease: an independent component analysis. J Neurosci. 2006 Oct 4;26(40):10222-31. PubMed.
Primary Papers
- Parra MA, Sala SD, Abrahams S, Logie RH, Méndez LG, Lopera F. Specific deficit of colour-colour short-term memory binding in sporadic and familial Alzheimer's disease. Neuropsychologia. 2011 Jun;49(7):1943-52. PubMed.
- Acosta-Baena N, Sepulveda-Falla D, Lopera-Gómez CM, Jaramillo-Elorza MC, Moreno S, Aguirre-Acevedo DC, Saldarriaga A, Lopera F. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer's disease: a retrospective cohort study. Lancet Neurol. 2011 Mar;10(3):213-20. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.