San Francisco: Making Tau Toxic—Post-translational Changes Galore
Quick Links
Given its correlation with Alzheimer’s disease progression, tau hyperphosphorylation has long commanded the field’s attention. Many labs have labored to identify disease-relevant phosphorylation sites on this microtubule-stabilizing protein, as well as the kinases and phosphatases responsible for those changes. Recent studies have shifted the focus, however. Some have uncovered acetylation, an altogether different post-translational modification, as potentially important for tau toxicity. Others are turning up potential targets for caspase-cleaved forms of pathological tau. These were among the topics scientists explored at a 28-30 March 2011 workshop, “Tau and Tauopathies: Pathogenic Mechanisms,” hosted by the Gladstone Institute of Neurological Disease in San Francisco and co-sponsored by the German Center for Neurodegenerative Diseases (DZNE) (see Part 1).
Acetylation—The New Force on the Block
Last year, researchers led by Li Gan at the Gladstone Institute reported that tau can be acetylated, potentially as a means of preventing its clearance by the ubiquitin-proteasomal system (ARF related news story on Min et al., 2010). Another study published earlier this year confirmed that tau is acetylated in cells, mice, and several human tauopathies. That report—by Virginia Lee and colleagues at the University of the Pennsylvania School of Medicine in Philadelphia—also confirmed that acetylation makes tau resistant to degradation and further suggested that acetylated tau is prone to fibrillization (ARF related news story on Cohen et al., 2011). First author Todd Cohen presented these data in San Francisco.
The data from these two labs suggest that reducing tau acetylation could be beneficial for treating tauopathies. In San Francisco, Gan said she wants to determine which acetylation sites are most relevant to AD; toward that end, efforts to generate monoclonal antibodies to specific epitopes are underway. The researchers are also making tauopathy mice with conditional deletion of SIRT1, an enzyme that can deacetylate tau. These tools should help address how acetylation affects tau function, Gan said. For their part, Cohen and colleagues have made an antibody that recognizes tau acetylated at K280, a site in the microtubule-binding region that is linked to frontotemporal dementia with parkinsonism-17 (FTDP-17). Deletion of this very lysine produces an aggregation-prone form of tau that causes synaptic and cognitive impairments when overexpressed in mice (Mocanu et al., 2008; Sydow et al., 2011). The UPenn researchers have confirmed that the antibody is sensitive and site-specific, and used it to examine acetylation of tau at K280 in several tau transgenic mouse models, and in postmortem brain tissue from AD patients (see below). Staining was low in hippocampal sections of PS19 mice carrying the P301S tau mutation, but much higher in PS19/PDAPP bigenic mice, Cohen reported.
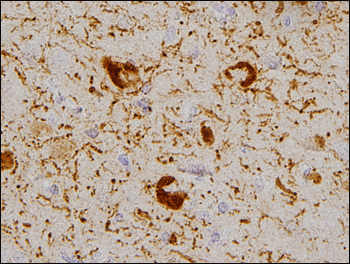
Tau protein acetylated at K280 shows up in neurofibrillary tangles from brains of patients with Alzheimer's disease. Image credit: Todd Cohen
“These are interesting new developments,” noted Eckhard Mandelkow of the DZNE in an e-mail to ARF. “However, since tau is an open, natively unfolded protein, any change in cellular activity will likely have some post-translational impact on tau. As in the extensively documented case of phosphorylation, it will be difficult to prove that acetylation modifications are causative, rather than correlative.”
Perhaps the same can be said for other pathological forms of tau, where research is just beginning to establish links with cellular pathways. Gail Johnson of the University of Rochester, New York, spoke about her lab’s recent studies with a caspase-cleaved tau fragment (tau-D421) that accumulates in the brains of AD patients and is neurotoxic in a fly model for AD and related tauopathies (Khurana et al., 2010). Using mouse cortical cells with doxycycline-inducible expression of D421 truncated tau, or full-length tau as a control, the researchers determined that truncated tau was more resistant to autophagic degradation (Dolan and Johnson, 2010). Taking a page from other work suggesting mitochondria as a potential target for pathological tau (see review by Eckert et al., 2010), the Rochester team found that turning on tau-D421 expression in cortical neurons boded badly for their energy-producing organelles. Compared to cells expressing full-length tau, tau-D421-expressing cells had more fragmented mitochondria and membrane damage, as well as higher oxidative stress levels (Quintanilla et al., 2009). In experiments measuring oxygen consumption rates of cells in 24-well plates, tau-D421-expressing neurons had abnormally low basal mitochondrial respiration and respiratory reserve capacity, Johnson reported. She concluded with fresh-off-the-bench Westerns showing that caspase-cleaved tau, but not wild-type tau, interacts with mitofusin 2 (Mfn2). Among other functions, Mfn2 regulates mitochondrial transport and is a primary gene mutated in Charcot-Marie-Tooth-type 2A neuropathy. Johnson’s preliminary data suggest Mfn2 as a potential target for pathological tau.
Phosphorylation—So Familiar, So Mysterious
In addition to the new data on post-translationally modified and caspase-cleaved forms of tau, the conference included several talks on the historic tau alteration—phosphorylation—and its role in neurodegeneration. A pioneer in this research area, Khalid Iqbal, New York State Institute, recapped recent data solidifying the long-held claim that this modification is a key means for making tau troublesome. Previously, he and colleagues determined that protein phosphatase 2A (PP2A) accounts for about 70 percent of total tau phosphatase activity in the human brain (Liu et al., 2005), and that AD patients have decreased activity of PP2A and PP1 in their brains (Gong et al., 1993). Inhibitor-2 of PP2A, also called SET, moves from the neuronal nucleus to the cytoplasm, where it settles within neurofibrillary tangles in the brains of people with AD (Tanimukai et al., 2005).
More recently, Iqbal’s group expressed the C-terminal fragment of SET in the brains of rats using adeno-associated viruses. This caused abnormal tau phosphorylation, neurodegeneration, and cognitive deficits, solidifying the idea that phosphorylation of tau does, in fact, promote its neurotoxicity (Wang et al., 2010).
Continuing the phosphorylation theme, research led by Akihiko Takashima at the RIKEN Brain Science Institute in Wako City, Japan, focused on glycogen synthase kinase 3 (GSK-3), which phosphorylates tau, and evidence for its necessity in AD pathogenesis (see review by Takashima, 2006). In San Francisco, Takashima discussed a simple model in which AD is triggered by Aβ-related events and/or by aging factors that induce neurofibrillary tangles. He presented data suggesting that tau and GSK-3β deficiencies do not give rise to detectable symptoms early on, but cause problems as the animals age. In their analyses of GSK-3β and tau knockout mice, Takashima and colleagues found that younger animals did fine on spatial learning and memory tests, but showed deficiencies by 22-23 months of age. When the researchers tried to induce long-term depression (LTD), which normally activates GSK-3β, deficits showed up in both young and old GSK-3β and tau knockout mice. These data suggest that mice become more sensitive to LTD deficits as they age, Takashima said.
However, despite the long-standing focus on tau hyperphosphorylation, scientists do not yet have a handle on how this modification actually gives rise to disease (see review by Takashima, 2010).
Is phosphorylated tau functionally important? This question guides much of the work in the lab of Gloria Lee, University of Iowa, Iowa City, who presented some of her studies on tau as a signal transduction protein. Previously, Lee and coworkers had shown that tau interacts with the tyrosine kinases Src and Fyn in vitro (see review by Lee, 2005). Subsequent work, including analyses of tauopathy mice, suggests that some of these associations may be relevant to neurodegenerative disease (Bhaskar et al., 2005; Bhaskar et al., 2010).
Probing whether tyrosine-phosphorylated tau has a physiological role, Lee and colleagues found clues in a previously published microarray analysis of brain tissue from tau-deficient mice (Oyama et al., 2004). Cytoskeletal factors were notably absent from that paper’s top 10 list of mRNAs regulated by tau, suggesting that tau’s major function may not be cytoskeletal, Lee said. Instead, transcripts encoding the transcription factor FosB took the first two spots on that list. Lee’s team explored whether tau influences the signaling pathway leading to FosB expression by monitoring activation of AP-1—a protein that complexes with Fos to bind DNA—in tau-overexpressing and tau-deleted cell lines stimulated by nerve growth factor. The researchers found that AP-1 activity (judged by MAP kinase activation) was low in the tau-deficient cells, and that introducing wild-type tau into the cells invigorated the pathway. The effect held even when they put in a tau mutant that cannot bind microtubules, suggesting that “whatever tau is doing here, it doesn’t have to do with microtubules,” Lee said.
The scientists determined that a specific phosphorylation site on tau, Thr231, is critical for the protein’s ability to promote MAPK activation, and studies are underway to identify proteins that interact with tau further upstream in the AP-1/MAPK pathway, Lee said. In her working model, the road to neurodegeneration begins with abnormal phosphorylation of tau, which causes changes in cell signaling that lead to loss of cell cycle control. To see whether tau might, in fact, have a role in cell division, Lee’s lab is doing experiments in human prostate cancer cells, which, unlike mice, express all six tau isoforms that appear in the brain (see review by Souter and Lee, 2010).—Esther Landhuis.
This is Part 2 of a four-part series. See also Part 1, Part 3, Part 4. Read the PDF of the entire series.
References
News Citations
- San Francisco: Gladstone Institute Hosts Tau Powwow
- Tau Timing: New Findings on Disease Progression, Clearance
- Tau Modification—Move Over Phosphate, Make Room for Acetylation
- San Francisco: Is Tau Reduction a Good Thing?
- San Francisco: Tau—Time to Shine as Therapeutic Target?
Paper Citations
- Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010 Sep 23;67(6):953-66. PubMed.
- Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VM. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. PubMed.
- Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, Petrova O, Schönig K, Bujard H, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008 Jan 16;28(3):737-48. PubMed.
- Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, D'Hooge R, Alzheimer C, Mandelkow EM. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci. 2011 Feb 16;31(7):2511-25. PubMed.
- Khurana V, Elson-Schwab I, Fulga TA, Sharp KA, Loewen CA, Mulkearns E, Tyynelä J, Scherzer CR, Feany MB. Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet. 2010 Jul;6(7):e1001026. PubMed.
- Dolan PJ, Johnson GV. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J Biol Chem. 2010 Jul 16;285(29):21978-87. PubMed.
- Eckert A, Schulz KL, Rhein V, Götz J. Convergence of amyloid-beta and tau pathologies on mitochondria in vivo. Mol Neurobiol. 2010 Jun;41(2-3):107-14. PubMed.
- Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GV. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J Biol Chem. 2009 Jul 10;284(28):18754-66. PubMed.
- Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005 Oct;22(8):1942-50. PubMed.
- Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993 Sep;61(3):921-7. PubMed.
- Tanimukai H, Grundke-Iqbal I, Iqbal K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer's disease. Am J Pathol. 2005 Jun;166(6):1761-71. PubMed.
- Wang X, Blanchard J, Kohlbrenner E, Clement N, Linden RM, Radu A, Grundke-Iqbal I, Iqbal K. The carboxy-terminal fragment of inhibitor-2 of protein phosphatase-2A induces Alzheimer disease pathology and cognitive impairment. FASEB J. 2010 Nov;24(11):4420-32. PubMed.
- Takashima A. GSK-3 is essential in the pathogenesis of Alzheimer's disease. J Alzheimers Dis. 2006;9(3 Suppl):309-17. PubMed.
- Takashima A. Tau aggregation is a therapeutic target for Alzheimer's disease. Curr Alzheimer Res. 2010 Dec;7(8):665-9. PubMed.
- Lee G. Tau and src family tyrosine kinases. Biochim Biophys Acta. 2005 Jan 3;1739(2-3):323-30. PubMed.
- Bhaskar K, Yen SH, Lee G. Disease-related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem. 2005 Oct 21;280(42):35119-25. PubMed.
- Bhaskar K, Hobbs GA, Yen SH, Lee G. Tyrosine phosphorylation of tau accompanies disease progression in transgenic mouse models of tauopathy. Neuropathol Appl Neurobiol. 2010 Oct;36(6):462-77. PubMed.
- Oyama F, Kotliarova S, Harada A, Ito M, Miyazaki H, Ueyama Y, Hirokawa N, Nukina N, Ihara Y. Gem GTPase and tau: morphological changes induced by gem GTPase in cho cells are antagonized by tau. J Biol Chem. 2004 Jun 25;279(26):27272-7. PubMed.
- Souter S, Lee G. Tubulin-independent tau in Alzheimer's disease and cancer: implications for disease pathogenesis and treatment. Curr Alzheimer Res. 2010 Dec;7(8):697-707. PubMed.
Other Citations
External Citations
Further Reading
Papers
- Khurana V, Elson-Schwab I, Fulga TA, Sharp KA, Loewen CA, Mulkearns E, Tyynelä J, Scherzer CR, Feany MB. Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet. 2010 Jul;6(7):e1001026. PubMed.
- Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VM. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. PubMed.
- Wang X, Blanchard J, Kohlbrenner E, Clement N, Linden RM, Radu A, Grundke-Iqbal I, Iqbal K. The carboxy-terminal fragment of inhibitor-2 of protein phosphatase-2A induces Alzheimer disease pathology and cognitive impairment. FASEB J. 2010 Nov;24(11):4420-32. PubMed.
- Quintanilla RA, Matthews-Roberson TA, Dolan PJ, Johnson GV. Caspase-cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J Biol Chem. 2009 Jul 10;284(28):18754-66. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.