San Diego: “Calcinists” See Years of Compensation Prior to Alzheimer’s
Quick Links
If our summary on Aβ oligomers from the recent conference of the Society for Neuroscience delivered an overdose of news on synaptotoxic Aβ, take this story as an antidote. Held 3-7 November in San Diego, California, the conference featured a wealth of presentations to reflect a broadening knowledge base about this disease, and the role of presenilin and calcium in AD pathogenesis is one of the exciting examples. Perturbations to the neuronal calcium balance in both aging and AD have been described for some years and have led to a calcium hypothesis of brain aging and dementia. What is new is an evolving concept tying intraneuronal calcium increases in important functional ways to known players in AD risk and pathogenesis, that is, to presenilin (PS) mutations, APP mutations, ApoE4, synaptic dysfunction, apoptosis. In particular, new research is trying to assess these links in model systems at early stages, long before visible pathologies have formed and could confound results. In essence, the new calcium research is reaching for a grasp of how subtle disease-promoting changes play against compensatory mechanisms that the neuron deploys to maintain homeostasis. “Calcinists” view the emergence of amyloid and tau pathology as the result of a breakdown of this compensation. As a conference offering, below is a summary of one such presentation by Grace (aka Beth) Stutzmann at Rosalind Franklin University of Medicine and Science, North Chicago, Illinois. For a broader view, read her eloquent review, published last month (Stutzmann, 2007).
In her talk, Stutzmann focused on the role of presenilin (PS) mutations and the ryanodine receptor. Unlike presenilin, this receptor is not a household term among “Alzheimerologists.” In a nutshell, Stutzmann proposed that, in mice expressing mutant human presenilin 1, the ryanodine receptor is hyperexcitable. Prone to dumping large amounts of Ca2+ from ER stores preferentially into distal dendrites and synaptic spines, it tends to end up dampening the activity of those synapses.
For background, Stutzmann first reminded the audience that neurons maintain a deep valley of nanomolar Ca2+ in the cytosol vis-à-vis millimolar Ca2+ outside the cell membrane on one side, and high micromolar Ca2+ inside the endoplasmic reticulum (ER) on the other side. Multiple different calcium channels and pumps in the cell membrane and the ER membrane are necessary to keep Ca2+ distributed across these gradients. In previous work, Stutzmann had set up a system that combines whole-cell patch clamp recordings with 2-photon imaging of calcium flows. This is done in thick, 300-micron cortical slabs from various mouse models. She routinely isolates three types of calcium response:
- One through the plasma membrane (where action potentials trigger calcium flow through voltage-gated Ca2+ channels)
- One through the IP3 receptor in the ER membrane (triggered by photolysis of caged IP3)
- One through the lesser-known ryanodine receptor (using its agonist caffeine and its blocker dantrolene)
Together, this experimental setup serves as a “holistic” system of assessing neurophysiologic consequences of specific calcium changes, Stutzmann said.
Stutzmann had shown before that neurons from triple transgenic mice created in Frank LaFerla’s lab, where she had been a postdoctoral fellow, readily show much stronger calcium outflows from the ER than do those of non-transgenic mice. By comparing this strain with a different model overexpressing only mutant PS1, and with an APP/tau double transgenic, she attributed this defect in calcium management to presenilin. The present focus on the ryanodine receptor arose with the twin observations that PS1-transgenic mice have elevated levels of ryanodine receptor protein, and that blocking this receptor normalized excessive calcium outflow from the ER. In the presenilin transgenic mice, the ryanodine receptor mediated the majority of this particular calcium flow, up from 20 percent in normal mice to 70 percent in the mutant mice (Stutzmann et al., 2006).
At the conference, Stutzmann showed newer data suggesting that the ER within the distal dendrites and even spines releases the greatest relative increases in ryanodine-triggered calcium in the PS1-mutant mice; it is not the ER around the nucleus and cell body where most textbook diagrams depict it to be. (The ER is known to extend into dendrites and even into spine heads; see picture below.) Those distal ER tips show little ryanodine receptor-mediated Ca2+ release in non-transgenic mice, but in PS1-mutant they increase this flow dramatically by 10-fold. By contrast, the ER in the cell body about doubled its ryanodine receptor-mediated outflow. This suggests that the synaptic areas of the ER are functionally separate from what is going on in the cell body, Stutzmann said.
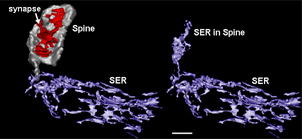
Three-dimensional Reconstruction of the Smooth ER (Purple) in a Rat Hippocampal CA1 Dendritic Segment
The left side shows that the ER in the dendrite is contiguous with the ER entering the thin neck of a dendritic spine (grey). The smooth ER in the head of the spine (right) is thought to provide synapse-specific regulation of calcium release, and modulate incoming synaptic signals (Reproduced from SynapseWeb; Spacek and Harris, 1997).
Does this mean anything to cortical neurons? These cells integrate multiple signals, and then generate a summed reaction. Stutzmann tried to model this behavior by assessing calcium release from the ER in response to either synaptic stimulation or ryanodine receptor activation alone, or both in combination. In the latter experiment, she saw a supra-additive depletion of ER calcium stores in the PS1-transgenic mice. Further experiments showed that this leads to greater membrane hyperpolarization. And this, in turn, hampered the synapses’ ability to generate trains of action potentials in response to electrical stimulation, essentially making the neuron less excitable (Stutzmann et al., 2007). “A main point here is that, depending on where it is dumped from the ER, this extra calcium will have very different functional consequences for the neuron,” Stutzmann said. (Links between ryanodine receptor function and mitochondria and apoptosis, for example, have been reported, as well.)
The existing literature indicates no change in synaptic transmission or synaptic plasticity in the triple transgenic mice at the young ages of 4 to 6 weeks, and Stutzmann’s group confirmed these findings. Yet she felt she had not looked hard enough. Maybe there was a compensatory effort hidden behind the curtain of this normal-looking output? “Calcium plays such a big role in synaptic transmission and plasticity, I don’t see how you can mess with it on a major scale and not affect those functions,” Stutzmann added. To peek behind the curtain, she blocked the ryanodine receptor with the drug dantrolene. In non-transgenic mice, this did away with about half the LTP output, but in PS1-transgenic mice it abolished LTP completely.
To Stutzmann’s mind, this suggests that the ryanodine receptor calcium stores in the dendritic tips of the ER contribute to synaptic function quite differently when there is a PS1 mutation. That the neurons at this age still manage to generate normal-looking LTP means that they are compensating, she said. This happens before any AD-like histopathology can be detected. Speculating about PS1-mutant FAD, this could imply that people are born with these different calcium dynamics and compensate well, until years of effort to maintain homeostasis exhaust the neuron and it switches from compensation to pathology. One future experiment would be to slightly dial down the ryanodine receptor over longer periods of time with drugs such as dantrolene, and ask whether that can keep AD-like pathology and behavioral deficits at bay in those models.
This summary covers but one area of active research on calcium flows in AD models. Effects of presenilin on ER calcium were established by a number of groups (Tu et al., 2006; Nelson et al., 2007; LaFerla, 2002), and the link between the ryanodine receptor, calcium, and AD is even older (Querfurth et al., 1998). Current questions focus on the relative contributions of the ryanodine versus the inositol trisphosphate IP3 receptor, as well as on how the two interact. This writer invites comments on these issues to broaden the discussion.—Gabrielle Strobel.
References
Paper Citations
- Stutzmann GE. The pathogenesis of Alzheimers disease is it a lifelong "calciumopathy"?. Neuroscientist. 2007 Oct;13(5):546-59. PubMed.
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci. 2006 May 10;26(19):5180-9. PubMed.
- Spacek J, Harris KM. Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci. 1997 Jan 1;17(1):190-203. PubMed.
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Parker I, Laferla F. Enhanced ryanodine-mediated calcium release in mutant PS1-expressing Alzheimer's mouse models. Ann N Y Acad Sci. 2007 Feb;1097:265-77. PubMed.
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006 Sep 8;126(5):981-93. PubMed.
- Nelson O, Tu H, Lei T, Bentahir M, de Strooper B, Bezprozvanny I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest. 2007 May;117(5):1230-9. Epub 2007 Apr 12 PubMed.
- Laferla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002 Nov;3(11):862-72. PubMed.
- Querfurth HW, Haughey NJ, Greenway SC, Yacono PW, Golan DE, Geiger JD. Expression of ryanodine receptors in human embryonic kidney (HEK293) cells. Biochem J. 1998 Aug 15;334 ( Pt 1):79-86. PubMed.
External Citations
Further Reading
Papers
- Rybalchenko V, Hwang SY, Rybalchenko N, Koulen P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int J Biochem Cell Biol. 2008;40(1):84-97. PubMed.
- Lee SY, Hwang DY, Kim YK, Lee JW, Shin IC, Oh KW, Lee MK, Lim JS, Yoon DY, Hwang SJ, Hong JT. PS2 mutation increases neuronal cell vulnerability to neurotoxicants through activation of caspase-3 by enhancing of ryanodine receptor-mediated calcium release. FASEB J. 2006 Jan;20(1):151-3. PubMed.
- Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007 Jun;6(3):337-50. PubMed.
- Thibault O, Gant JC, Landfield PW. Expansion of the calcium hypothesis of brain aging and Alzheimer's disease: minding the store. Aging Cell. 2007 Jun;6(3):307-17. PubMed.
- Smith IF, Hitt B, Green KN, Oddo S, Laferla FM. Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer's disease. J Neurochem. 2005 Sep;94(6):1711-8. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Kentucky
This article by Gabrielle Strobel provides an excellent review of recent research advances analyzing the nature and timing of calcium dysregulation in brain neurons of Alzheimer disease (AD) model mice, particularly highlighting the elegant imaging and electrophysiological studies of ryanodine receptor (RyRs) function by Grace Stutzmann and her colleagues. This work is clearly exciting and promising. However, it also seems important to view it in the context of dynamic age-dependent changes and potential interactions with other pathways in Ca2+ dysregulation. The effect of presenilin mutations on Ca2+ release from RyRs is likely to reflect only one stage of a complex cascade of abnormal Ca2+ signaling that, as the article noted, may begin well before hallmark pathology appears, perhaps during normal aging.
For example, early in the development of the Ca2+ hypothesis, we found evidence of increased Ca2+ signaling and voltage-gated Ca2+ influx in hippocampal neurons during normal aging in rats (Landfield and Pitler, 1984). This observation has been extended since by imaging and single channel recording studies (Thibault et al., 1996; Thibault et al., 2001; Hemond and Jaffe, 2005), and such increased Ca2+-dependent processes have been shown to be relevant to age-associated learning deficits (Disterhoft et al., 2004). Furthermore, a recent age course study found that onset of increased Ca2+ release from RyRs with normal aging may underlie wide disturbances in Ca2+-dependent processes (Gant et al., 2006), conceivably, in tandem with or in response to increased influx through voltage-gated Ca2+ channels (Thibault et al., 2007).
As Stutzmann has pointed out, presenilin mutations alone, although they account for the effect on RyR Ca2+ release, are not sufficient to induce pathological hallmarks of AD or neurodegeneration. Moreover, gene mutations account for only a few percent of AD cases. For the vast majority of sporadic cases, then, AD is overwhelmingly a disease of aging. Thus, if Ca2+ dyshomeostasis is indeed a key player in the transition from normal brain aging to AD, our understanding of the role of “calcinopathies” in AD will clearly be informed by unraveling the nature of multiple Ca2+ disturbances at the intersection between normal brain aging and AD.
References:
Landfield PW, Pitler TA. Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science. 1984 Nov 30;226(4678):1089-92. PubMed.
Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996 May 17;272(5264):1017-20. PubMed.
Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001 Dec 15;21(24):9744-56. PubMed.
Hemond P, Jaffe DB. Caloric restriction prevents aging-associated changes in spike-mediated Ca2+ accumulation and the slow afterhyperpolarization in hippocampal CA1 pyramidal neurons. Neuroscience. 2005;135(2):413-20. PubMed.
Disterhoft JF, Wu WW, Ohno M. Biophysical alterations of hippocampal pyramidal neurons in learning, ageing and Alzheimer's disease. Ageing Res Rev. 2004 Nov;3(4):383-406. PubMed.
Gant JC, Sama MM, Landfield PW, Thibault O. Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci. 2006 Mar 29;26(13):3482-90. PubMed.
Thibault O, Gant JC, Landfield PW. Expansion of the calcium hypothesis of brain aging and Alzheimer's disease: minding the store. Aging Cell. 2007 Jun;6(3):307-17. PubMed.
Make a Comment
To make a comment you must login or register.