Systemic Inflammation: A Driver of Neurodegenerative Disease?
Quick Links
Under healthy conditions, microglia look placid. They sit evenly spaced throughout the brain, processes extended, quietly doing their job of scanning for debris. When disease kicks in, these calm cells can transmogrify and end up doing more harm than good. As discussed at “Neuroinflammation in Diseases of the Central Nervous System,” a Keystone meeting held January 25-30 in Taos, New Mexico, the rabble-rousing signals that fire up microglia are not confined to the brain but also come from “below the neck,” said Hugh Perry of the University of Southampton in England. Whether triggered by acute infections or chronic disease, systemic inflammation may amplify microglial responses and exacerbate neurodegeneration, according to researchers at the meeting. They proposed ways to slow disease progression by soothing systemic inflammation.
The human body accumulates inflammatory battle scars as we age, whether through repeated assaults by microbial infections or chronic inflammatory diseases such as diabetes or atherosclerosis. Considering how systemic inflammation might alter the course of neurodegeneration is thus crucial, Perry said. “Old brains are attached to old bodies, and bodies tend to accumulate a lot of pathology over the years,” he said. At the meeting, Perry reported that this pathology primes microglia, making them prone to overreactions that could exacerbate neurodegeneration.
More than a dozen years ago, Perry and colleagues used a mouse model of prion disease to establish the idea that microglia can be primed. They found that microglia in prion-infected mice exhibited a state of heightened activation, and that the cells responded with extra gusto—pumping out pro-inflammatory cytokines and accelerating neurodegeneration—when the researchers subsequently challenged the mice systemically with lipopolysaccharide (see Combrinick et al., 2002).
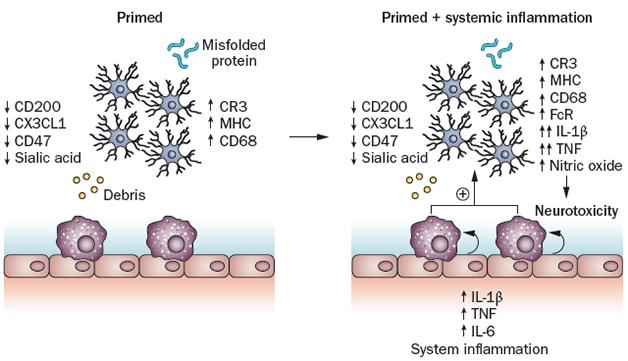
Priming the Inflammatory Pump. In response to misfolded proteins or debris from neurodegeneration, microglia (blue cells) ramp up production of some proteins (left panel). When systemic inflammation strikes, perivascular macrophages (purple) and endothelial cells kick the primed microglia into overdrive (right panel). [Image courtesy of Perry and Holmes, Nature Reviews Neurology, 2014.]
The group has since reported that inflammation from inside or outside the brain can prime microglia (see Perry and Holmes, 2014). In one study, systemic infection with salmonella stimulated microglia to secrete pro-inflammatory cytokines and express surface activation markers for several weeks. Endothelial cells lining the blood-brain barrier (BBB) also responded to the systemic alarm by expressing adhesion molecules. Subsequent injection of lipopolysaccharide into the brains of primed mice triggered much higher levels of microglial activation than in mice that hadn’t been infected (see Püntener et al., 2012).
Perry hypothesized that similar triggering scenarios must happen in the human brain throughout life, and that this would affect the progression of neurodegeneration. In collaboration with Clive Holmes at the University of Southampton, the researchers tracked cognitive symptoms in 300 people with AD over a six-month period. They found that cognition in patients who came down with a systemic infection declined twice as fast as it did in those who didn’t. People with chronic inflammation, as measured by high baseline levels of the proinflammatory cytokine tumor necrosis factor alpha (TNFα), declined at quadruple the rate of those without (see Holmes et al., 2009).
Based on this study, Holmes and Perry hypothesized that blocking TNFα would slow disease. At Keystone, Perry presented the results of a small pilot study that tested this. Over six months, the researchers treated 20 AD patients with weekly subcutaneous injections of etanercept, a decoy receptor that binds TNFα. The researchers monitored the patients’ performance on neuropsychiatric tests and activities of daily living. They found that while 20 other AD patients getting a placebo tended to decline in these measures over the study period, those taking the TNFa blocker stabilized. “As a pilot study, it shows you that the arrow points in the right direction,” Perry said. He stressed that larger studies will be needed to see if the effect holds. The results offered a glimmer of hope that targeting inflammation could have therapeutic potential, especially following disappointing results of non-steroidal anti-inflammatory drugs (NSAIDs) for AD (see May 2008 news).
Malu Tansey of Emory University in Atlanta asked Perry if he thought etanercept could affect AD progression by working systemically, or by eking into the brain in small amounts. Perry said there was scant evidence that the drug enters the brain at all, so it was likelier that any effects on AD would occur by quelling systemic inflammation.
Kathryn MacPherson, a graduate student in Tansey’s lab, presented initial findings from a study of a more selective TNFα inhibitor, XPro1595. TNFα exists in two forms that have opposing effects. A soluble form triggers pro-inflammatory responses that promote chronic inflammation and cell death, whereas the transmembrane version of the protein facilitates neuroprotective responses (see Montgomery and Bowers, 2011). Unlike etanercept, which blocks both forms of TNFα, XPro1595 only blocks soluble TNFα. The inhibitor also enters the brain.
MacPherson reported that 5xFAD mice display age-dependent brain inflammation. Treating the mice with XPro1595 reduced the number of activated, CD45-hi myeloid-derived cells (presumably infiltrating monocytes) in the brain, and also altered T-cell populations there. While the inhibitor’s effects on plaques have not yet been measured, MacPherson reported that in studies performed in collaboration with Chris Norris at the University of Kentucky, it rescued deficits in synaptic strength and long-term potentiation in 7-month-old 5xFAD mice. The researchers did not observe an elevation in systemic inflammation in 5xFAD mice as compared to non-transgenic mice, suggesting that XPro1595 may work by calming inflammation in the brain. To measure the drug’s effects in a situation more akin to aging people with chronic systemic inflammation, MacPherson plans to investigate the effects of the inhibitor in AD mice on a high-fat or high-sugar diet. FPRT, a small biotech company, is developing the drug for the treatment of neurodegenerative diseases, and it reduces cell loss in a rat model of Parkinson's (see Barnum et al., 2014).
Glenn Rall of Fox Chase Cancer Center in Philadelphia presented another example of cross-talk between immune responses in the brain and the rest of the body. Rall detailed how T cells responding to a virus in the periphery can move into the brain and wreak havoc there. He studied mice systemically infected with lymphocytic choriomeningitis virus (LCMV), which evokes a strong CD8+ T cell response but does not infect the brain. While this virus raged, he injected mice intracranially with a measles virus that only infects neurons in the CNS. The mice survived infection by either of the two viruses alone without even getting sick, but co-infection killed half of them. The mice died because LCMV-specific CD8+ T cells from the periphery infiltrated the brain, where they triggered deadly edema. These cells stayed out of the brain of mice infected only with LCMV, suggesting that the CNS measles infection set the stage for the disastrous recruitment (see Matullo et al., 2011). Rall found that other virus combinations, such as systemic ectromelia combined with CNS-restricted polio, triggered a similar infiltration of T cells.
Could a systemic infection set the stage for T-cell incursion into the brain even after the illness has passed? To test this, Rall infected mice systemically with LCMV, waited a month (long after the virus was cleared), and then infected their CNS with measles. These mice became ill, unlike controls that received only the measles virus. Rall detected T-cell infiltration in this model as well, but has not yet confirmed those T cells were specific to LCMV.
Although T cells were the agents of destruction in Rall’s co-infection model, he plans to investigate whether microglia or infiltrating macrophages were instigators. It’s possible that primed macrophages beckoned the T cells into the brain, Rall said.
Perry was fascinated by the idea that CNS infection opened the brain to infiltration by peripheral immune cells. He said that in his salmonella infection model, peripheral T cells also infiltrated the brain, even though the BBB remained intact. He speculated that activated endothelial cells within the BBB somehow facilitated this recruitment.
Along those lines, Richard Daneman of the University of California, San Diego, focused on structural changes that weakened the blood-brain barrier in disease. Although endothelial cells that make up the BBB are akin to those that line other vessels in the body, BBB endothelial cells form a much tighter barrier. In a previous study, Daneman and colleagues found that BBB endothelial cells maintain this barrier in response to signals from pericytes. The latter lie on the parenchymal side of the BBB and communicate with brain-resident cells and the endothelium. Pericytes prevent BBB endothelial cells from expressing “leaky genes” expressed by endothelial cells in other parts of the body (see Daneman et al., 2010). At Keystone, Daneman reported that one of these genes, EHD4, regulates endothelial permeability. Normally, BBB endothelial cells do not express this vesicular protein, but in mouse models of stroke, multiple sclerosis, traumatic brain injury, and epilepsy, or in pericyte-deficient mice, they upregulate EHD4. When Daneman overexpressed EHD4 specifically in BBB endothelial cells, he observed that the protein localized within vesicles of the cells, where it facilitated the transcytosis of proteins normally barred from the brain, such as fibrinogen.
Daneman proposed that during disease, pericytes lose their connections with endothelial cells, lifting suppression of EHD4 and other genes that promote unintended transport across the BBB. Daneman has so far tested this theory in CNS disease models, but researchers at the meeting wondered if the same breakdown happens during systemic inflammatory diseases, such as lupus or atherosclerosis. If this were the case, targeting EHD4 and related pathways could offer a way to prevent barrier disruption in many diseases, Daneman said.
Would such barrier disruption promote the damaging cross-talk that Perry and Rall saw in their models of microglial priming and T-cell infiltration? Perry told Alzforum that so far, evidence suggests that the communication between the periphery and the brain occurs across an intact BBB in these models, although he offered that a transient breakdown in the BBB could facilitate enhanced infiltration of inflammatory mediators. Rall agreed. “A leaky barrier is indiscriminate in terms of what it allows into the brain,” he said. “I think these immune cells are crossing capillaries with an intact barrier, due to an activated endothelium.”
The integrity of the BBB, and how it relates to communication with the rest of the body during disease, remains an area of active debate. For example, at Keystone, Ryan Watts of Genentech in San Francisco suggested that the barrier remains intact in mouse models of AD, as well as in tau transgenic and ApoE-deficient mice. Watts measured transport of antibodies across the BBB in these mice and found it was no different from wild-type. Human disease may be a different story, however. Researchers recently reported subtle leaks in the hippocampus of older people and in people with mild cognitive impairment, though this finding has not yet been independently replicated (see Alzforum Webinar).
Whether the flow of immune communication is haphazard or carefully orchestrated, researchers seemed to agree that the brain and the body share the burden of inflammation during aging.—Jessica Shugart
References
News Citations
Webinar Citations
Paper Citations
- Combrinck MI, Perry VH, Cunningham C. Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience. 2002;112(1):7-11. PubMed.
- Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014 Apr;10(4):217-24. Epub 2014 Mar 18 PubMed.
- Püntener U, Booth SG, Perry VH, Teeling JL. Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J Neuroinflammation. 2012;9:146. PubMed.
- Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, Culliford D, Perry VH. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009 Sep 8;73(10):768-74. PubMed.
- Montgomery SL, Bowers WJ. Tumor Necrosis Factor-alpha and the Roles it Plays in Homeostatic and Degenerative Processes Within the Central Nervous System. J Neuroimmune Pharmacol. 2011 Jul 5; PubMed.
- Barnum CJ, Chen X, Chung J, Chang J, Williams M, Grigoryan N, Tesi RJ, Tansey MG. Peripheral administration of the selective inhibitor of soluble tumor necrosis factor (TNF) XPro®1595 attenuates nigral cell loss and glial activation in 6-OHDA hemiparkinsonian rats. J Parkinsons Dis. 2014;4(3):349-60. PubMed.
- Matullo CM, O'Regan KJ, Curtis M, Rall GF. CNS recruitment of CD8+ T lymphocytes specific for a peripheral virus infection triggers neuropathogenesis during polymicrobial challenge. PLoS Pathog. 2011 Dec;7(12):e1002462. Epub 2011 Dec 22 PubMed.
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010 Nov 25;468(7323):562-6. Epub 2010 Oct 13 PubMed.
Other Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
GSK Vaccines
Interesting concept indeed. It is now more evident that peripheral immune system misregulation plays a key role in Alzheimer’s disease. Just the number of genes involved in innate immunity identified by genome-wide associations studies is enough to give us evidence, or at least hints. It’s all about a balance between high inflammation, such as might result from a viral or bacterial infection (which seems to be detrimental to AD) and daily immune-surveillance, which might be beneficial for us to deal with protein aggregates or oligomers every day (tau, Aβ, prions, synuclein). Given that non-steroidal immunosuppressants failed (NSAID) and even exacerbated AD, I would also be cautious to blunt completely the TNF, such as with etanercept, since TNF is important for our daily immunosurveillance to control infections.
It is interesting once again that highly pyrogenic molecules and those that activate TNF, such as LPS, in peripheral blood are detrimental in amyloid accumulating models as demonstrated previously. (See previous Alzforum comments on Michaud et al., 2013). However, I would be cautious about jumping to conclusions. Mild peripheral "inflammation" NOT triggered by bacterial or viral infection or endotoxins might have a the opposite effect.
Immunology speaking, we should distinguish between inflammation versus controlled innate and adaptive immune response and not draw conclusions that peripheral cytokines such as TNF, or others, are always accelerating AD. Precise dose-response studies will need to be performed with such cytokines or inhibitors.
References:
Michaud JP, Hallé M, Lampron A, Thériault P, Préfontaine P, Filali M, Tribout-Jover P, Lanteigne AM, Jodoin R, Cluff C, Brichard V, Palmantier R, Pilorget A, Larocque D, Rivest S. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer's disease-related pathology. Proc Natl Acad Sci U S A. 2013 Jan 29;110(5):1941-6. PubMed.
Make a Comment
To make a comment you must login or register.