Lewy Pathology in DLB Spreads Fast, Maybe From the Nose
Quick Links
At the International Dementia With Lewy Body Conference, held December 1-4 in Fort Lauderdale, Florida, scientists suggested that concurrent amyloid pathology in DLB explains some of the overlap between DLB and Alzheimer’s disease (see Part 3 of this series). But they also reported indications that DLB is very much its own disease, and these came from DLB’s defining component, the α-synuclein pathology itself.
Glenda Halliday of University of New South Wales, Sydney, told the audience that while the knowledge base on Lewy pathology in aging humans comes largely from Parkinson’s, research on DLB is catching up and has found distinctive features in the appearance, timing, and spread of Lewy pathology. A typical Lewy body takes about a decade to grow and condense into its classic, structured form. In Parkinson’s disease, most Lewy bodies look that way. That is especially true of the older ones in the brainstem, which have had more time to mature than the more recent ones that arose later during the pathology’s gradual spread up to cortical regions. In contrast, Lewy bodies in DLB tend to look younger and less structured, Halliday said. They are also far more prevalent all over the cortex. They sweep through the brain faster, fueled perhaps by the co-occurrence of other pathologies. While people with PD survive on average 13 years after their diagnosis, DLB patients die well within a decade, some even as early as six months, after disease is apparent.
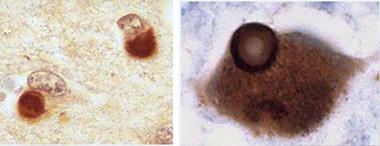
Two cortical neurons each contain a diffuse Lewy body as typically seen in DLB (left, x400). Mature Lewy body in a brainstem pigmented neuron, as typically seen in PD (right, x1000). This Lewy body is ringed by a halo. Both are stained for α-synuclein. [Courtesy of Glenda Halliday.]
“The tempo of DLB is faster, and no matter when a patient dies, even if it’s soon after their diagnosis, they tend to have Lewy pathology everywhere,” Halliday said.
In a given person, the amount of one pathology relates to the amount of another; in other words, people who have lots of Lewy bodies tend to have lots of cortical amyloid deposits, more abnormal neurites and cell loss in the hippocampus, more striatal amyloid deposits, and more atrophy. “All these measures correlate to the degree of Lewy pathology in DLB,” Halliday said.
It is not only the amount that is different, but apparently also the route the toxic proteins take. In DLB, the nose is increasingly attracting the attention of researchers as one possible explanation. Could it be that, at least in some DLB patients, α-synuclein deposition starts in the nose and spreads from there to other parts of the brain? The olfactory epithelium, which feeds into the olfactory bulb, is only a short distance away from the amydgala. The amygdala is a main hub of Lewy pathology in DLB, which in turn relays to many of the other affected areas. Thomas Beach of Banner Sun Health Research Institute in Sun City, Arizona, has long argued that a large proportion of DLB cases do not follow Braak’s brainstem-to-cortex progression of Lewy pathology. Beach and other researchers have implicated the olfactory mucosa as a possible entry zone instead (Beach et al., 2009; Funabe et al., 2012). From the nose, Lewy bodies could easily access the amygdala and then the brainstem, Beach said in Fort Lauderdale.
Tracing the route by which pathology marches through the brain invariably invokes the hypothesis of prion-like spread, and indeed this mechanistic discussion came up at the DLB conference, as well. Patrik Brundin of the Van Andel Research Institute in Grand Rapids, Michigan, drew much notice when he showed early data of a new animal model for a progressive spread of pathology starting in the nose. This model, a wild-type mouse, might be able to recapitulate a DLB- or PD-like disease.
Nolwen Rey in Brundin’s lab used sonicated α-synuclein fibrils in a collaboration with Kelvin Luk from Virginia Lee’s group at the University of Pennsylvania, Philadelphia, and injected them into the olfactory bulb of wild-type mice. Then she waited. Would they be taken up by neurons in the olfactory bulb? Would they recruit endogenous α-synuclein, seed aggregation, and lead to release of those aggregates from the neurons’ terminals in their projection area, the piriform cortex? Would they trigger this process to repeat itself in postsynaptic neurons there?
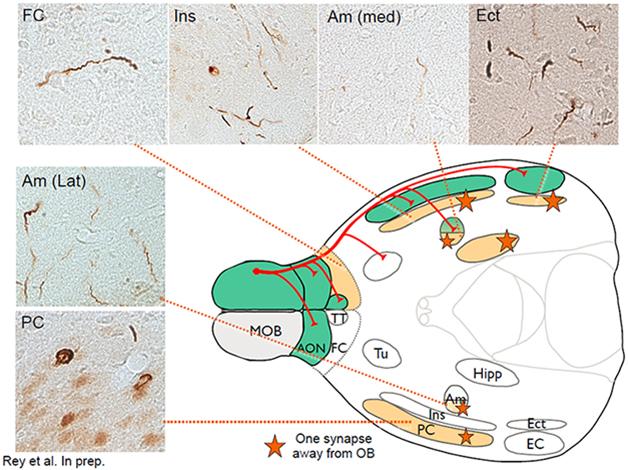
This schematic of the mouse brain depicts primary connections (red) going from the main olfactory bulb (MOB) to the piriform cortex (PC), the frontal cortex (FC), the amygdala (Am) and the entorhinal cortex (Ect). Green areas show inclusions stained with antibodies against phosphorylated α-synuclein (micrograph inserts) one month after fibril injection; orange areas, which involve traversing one synapse, three months after injection. [Courtesy of Nolwen Rey.]
Indeed, that is precisely what Rey is beginning to see, Brundin told the audience. This is ongoing work, but Brundin showed some slides indicating how, one month after the injection, α-synuclein aggregates appeared in immediate olfactory bulb projection areas. By three months, more aggregates showed up in the brain nuclei one synapse away (see image). By six months, aggregates appeared in additional areas two synapses removed from the olfactory bulb. A year after the injection, Rey saw α-synuclein deposits in about 60 brain areas and subregions that are anatomically connected, directly or indirectly, to the olfactory bulb. They are thioflavin-positive aggregates that share common markers with Lewy bodies, Brundin said. What’s more, the mice showed evidence of a progressive inability to distinguish scents, and to remember odors they had smelled before.
This spread is similar from mouse to mouse, and predictable in its timing. “There is not a lot of variability in this model,” Brundin told Alzforum, “It is very staged, and goes from the olfactory bulb to the amygdala. We have seen in some cases that it even reaches the brainstem.”
Brundin’s talk came after Virginia Lee of UPenn had set the stage for a discussion of the seeding hypothesis as an underlying mechanism. Lee focused on injections into the striatum, not the olfactory bulb. She recapped her lab’s prior work, which recently continued with a paper reporting a staged spread of α-synuclein pathology and neurodegeneration triggered by preformed fibrils in rats (Paumier et al., 2015; Nov 2012 news). “Our lab has done many injection experiments by now. We find that wherever we inject α-synuclein, its spread retraces that area’s anatomical connectome in exquisite detail,” Lee said. The seeding hypothesis is gaining ground across the field but is not uncontested (Apr 2014 news).
Brundin emphasized that templated seeding and spread starting in the nose could lead to both DLB or PD. Many people with either disease report having lost their sense of smell years before other symptoms of DLB or PD became apparent.
The idea that DLB can start in the nose is provocative but remains controversial, commented Bradley Boeve of the Mayo Clinic in Rochester, Minnesota. While he called Brundin’s model “compelling,” he also noted that many DLB and PD patients develop REM sleep disorder well over a decade before core DLB symptoms crop up. This, to Boeve and others, implies that pathology in the brainstem must start early. Researchers do not know yet if α-synuclein travels from the nose to the amygdala in some people and from the enteric system through the vagus nerve to the brainstem in other people. The clinical heterogeneity of α-synucleinopathies leaves room for multiple different paths in different people. In discussion, Brundin noted that because humans swallow their nasal secretions, both the nose and the gut in theory could be assaulted by the same environmental toxins, such as inhaled pesticides or metal fumes in welding.
“All hypotheses in DLB are difficult to test in humans without a more sensitive and specific biomarker,” Boeve said. Indeed, longitudinal biomarker studies are needed to chart the paths α-synucleinopathy takes and to understand the exact differences between DLB and PD. Until then, Brundin quipped, the relationship between the two evokes the Thai idiom “Same same, but different.”—Gabrielle Strobel
References
News Citations
- Dementia with Lewy Bodies: Sharper Image for a Formerly Blurry Disease
- Toxic Synuclein Corrupts Native in Wild-Type Mice
- Are Synuclein Seeds Non-Starters?
Paper Citations
- Beach TG, White CL, Hladik CL, Sabbagh MN, Connor DJ, Shill HA, Sue LI, Sasse J, Bachalakuri J, Henry-Watson J, Akiyama H, Adler CH, . Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol. 2009 Feb;117(2):169-74. PubMed.
- Funabe S, Takao M, Saito Y, Hatsuta H, Sugiyama M, Ito S, Kanemaru K, Sawabe M, Arai T, Mochizuki H, Hattori N, Murayama S. Neuropathologic analysis of Lewy-related α-synucleinopathy in olfactory mucosa. Neuropathology. 2012 Jun 4; PubMed.
- Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, Steece-Collier K, Kemp CJ, Celano S, Schulz E, Sandoval IM, Fleming S, Dirr E, Polinski NK, Trojanowski JQ, Lee VM, Sortwell CE. Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis. 2015 Oct;82:185-99. Epub 2015 Jun 17 PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.