11th ICFTD Meeting in Sydney Sorts Out Clinical Subtypes
Quick Links
Indigenous Australian people opened the 11th International Conference on Frontotemporal Dementia at the International Convention Center, Sydney, November 11–14. Koomurri performers welcomed more than 600 attendees from 37 countries. Chaired by University of Sydney’s Olivier Piguet on behalf of the local organizing committee, this latest installment of the biannual conference covered the gamut from basic biology to caregiving. A carers' day engaged affected families with personal experiences and updates on the latest research, clinical trials, care planning, and study initiatives. It reinforced, for researchers, the need to better diagnose, track, and treat the subtypes of FTLD. Scientists agreed that key to all three is nailing down what contributes to the cadre of protein pathologies that spread through specific cells and regions of the brain.
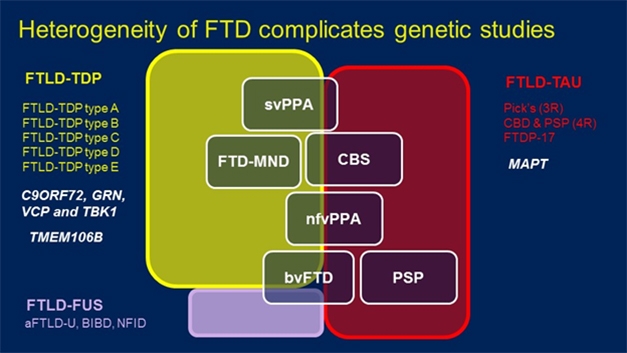
FTD Spectrum. Mutations in a variety of genes (white, left) can lead to pathological aggregates of TDP-43 (yellow), FUS (purple), and in the case of the MAPT gene, tau (red), underlie a handful of clinically distinguishable diseases (white boxes). [Courtesy of Rosa Rademakers.]
It’s complicated, confusing to many, and urgently requires clearer mapping of gene variants to pathologies and, especially, clinical phenotypes. For example, the field currently recognizes five different forms of FTLD based on the morphology of TDP-43 alone. Type A to E FTLD-TDP can manifest as behavioral variant FTD, FTD with motor neuron disease, semantic and non-fluent types of primary progressive aphasia, or corticobasal syndrome. Independently of TDP-43, some of these same clinical diagnoses are made when patients have inclusions of the microtubule-binding protein tau.
Hints at what causes FTD come from genetics. Variants in the tau gene, MAPT, cause the tauopathies, whereas variants in genes encoding four different proteins—C9ORF72, progranulin, valosin-containing protein, and TANK-binding kinase 1—lead to FTD marked by TDP43 inclusions. Could other variants influence what type of TDP43 morphology forms?
Rosa Rademakers at Mayo Clinic, Jacksonville, Florida, has collaborated with dozens of researchers in North America, Europe, and Australia to sequence whole genomes of people with pathologically confirmed FTD-TDP43. From 1,151 samples, the largest collection of FTLD brains to date, Rademakers has thus far sequenced genomes from 517 people with pathologically confirmed TDP43 disease who have no known pathogenic mutations in C9ORF72, PGRN, VCP, or TBK-1.
Comparing these genomes against those from 838 control samples from the Mayo Clinic, the geneticists uncovered two common single-nucleotide polymorphisms on chromosome 7q36 that reached genome-wide significance for association with FTLD. The SNPs lie in intron 1 of the gene for dipeptidyl peptidase like 6. DPP6 was previously linked to FTLD by Rita Cacace in Christine Van Broeckhoven’s group at the University of Antwerp (Sep 2016 conference news). Cacace found an inversion that disrupted the gene. With the new result, Rademakers considers DPP6 a likely candidate gene, though she agreed the data need replication—no small feat in a rare disease.
In the meantime, supporting data come from mice, which exhibit behavioral problems when they lack the gene. How DPP6 functions is unclear, but because it lacks a catalytic serine normally found in serine proteases, this peptidyl peptidase-like protein has no peptidase activity. Instead, DPP6 seems to modulate voltage-gated potassium channels needed for neurotransmission, said Rademakers.
Do any common variants account for specific TDP43 subtypes? Among 162 samples with Type A TDP43, Rademakers’ screen identified rs5848. This SNP lies downstream of the PGRN gene, and Rademakers and others have previously linked it to FTD (Cruts et al., 2006; Baker et al., 2006; Rademakers et al., 2008). The minor T allele seems to reduce progranulin expression. “If you have two copies, you have less progranulin, though not as little as if you have PGRN mutations,” said Rademakers. “This shows that progranulin deficiency may be a cause of FTD not only in people with PGRN mutations,” she said.
Analysis of 184 type B samples turned up a significant hit around UNC13A, another gene previously linked to FTD and ALS (Apr 2018 news; van Es et al., 2009). Scientists believe this protein regulates neurotransmitter release. Rademakers thought it telling that this association emerged from such a small sample set. Alas, in 143 Type C samples, the researchers were unable to find any variants linked with disease.
Next, Rademakers and colleagues looked for rare variants that might explain any form of TDP43 FTD. “We focused on loss-of-function mutations because we had the power to do that,” she said. This identified 61 genes which had at least three mutations among cases versus none in controls. One was the known FTD gene TBK1. ToppGene, an online platform that prioritizes candidates based on functional similarity, ranked three of the other 60 genes—DHX58, TRIM21, and IRF7—the highest. Their proteins perform similar roles to TBK1, being involved in innate immune signaling that leads to production of type I interferons. ToppGene further identified IRF3, IRF8, and NOD2, which are involved in the same pathway. Going back to the rare-variant analysis, Rademakers found one loss-of-function variant in each of these genes among the FTD cases, suggesting they may be candidate FTD genes, as well.
Another rare-variant came from a case-control study by Jennifer Yokoyama at the University of California, San Francisco. Among self-described non-Hispanic white volunteers, she looked for variants enriched in pathology-confirmed FTD cases compared with healthy age-matched controls. She focused on genes with four or more rare SNPs likely to have functional effects. Among 14,459 genes in an initial discovery data set of 62 cases and 2,599 controls, Yokoyama found that, on aggregate, nine variants of the MFSD8 gene occurred in 6.5 percent of FTLD patients but in only 0.4 percent of controls. Deeper analysis of 94 patients and 3,541 controls called the difference significant.
The MFSD8 gene encodes a lysosomal protein. Homozygous mutations at five different amino acids in MFSD8 cause the lipid storage disease late infantile neuronal ceroid lipofuscinosis (Stogmann et al., 2009). In this respect, MFSD8 is uncannily like progranulin, since homo- and heterozygous mutations in PGRN cause childhood lipid storage disorders and FTD, respectively.
Homozygous MFSD8 mutations deplete neurons in layer V of the frontal cortex, a region affected in FTD. In Sydney, Yokoyama reported that in heterozygous carriers with FTD, MFSD8 protein levels were elevated in the frontal gyrus, as were levels of lysosomal markers LAMP-2, CTSD, and LC3-II. This, too, is similar to what is seen in progranulin heterozygotes.
These changes suggest a deficit in lysosomal function, which was apparent in fibroblasts taken from carriers. Further, Yokoyama found that overexpressing one copy of the MFSD8 variant F379S in HEK cells caused small fragments of the protein to accumulate on the cell surface. “This suggests that altered trafficking of MFDS8 to lysosomes could result in reduced protein turnover, consistent with our observations of increased MFSD8 protein levels and reduced lysosomal functioning,” said Yokoyama. Some of this data appeared in the October 31 Acta Neuropathologica (Geier et al., 2018).
Other scientists at the meeting wondered if MFSD8 variants alter levels of progranulin, TDP43, or other proteins implicated in FTD. Yokoyama has not looked at this yet, but said that experiments with fibroblasts from MAPT and GRN mutations carriers hint as much.
Taking a different tack, Edward Lee, University of Pennsylvania, Philadelphia, reported that variants in C9ORF72 might predispose to corticobasal degeneration (CBD). While long hexanucleotide expansions in this gene cause FTD/ALS, Lee wondered if there were pleiotropic effects dependent on expansion length. There is precedent for this: For example, long polyglutamine expansions in ataxin-2 cause spinocerebellar ataxia, while shorter ones increase risk for ALS (Aug 2010 news). Moreover, earlier hints tying C9 expansions larger than 17 repeats to Parkinson’s disease were never proven, said Lee (Nuytemans et al., 2013; Nuytemans et al., 2014). He wondered if a PD mimic, such as CBD, might have been the real McCoy.
To test this idea, Lee analyzed samples from the largest cohort of autopsy-confirmed CBD cases to date, comprising tissue from 355 patients. He genotyped these for C9ORF72 using capillary electrophoresis to quantify expansions sizes, and compared them to expansions among 11,087 controls. Historically, expansion size has been difficult to determine by nucleotide amplification or Southern blot, because the sheer number of hexanucleotide repeats interferes.
Lee reported that repeats smaller than 17 were rarer in CBD patients than controls, but repeats larger than 17 were twice as likely to occur in CBD patients than controls. Why would these relatively small repeats cause CBD while repeats hundreds or even thousands long cause FTD/ALS? Lee believes it’s about gene expression. C9ORF72 has three isoforms, and in the brain samples, mRNA levels for the third isoform rose as expansions grew from 17 to 30 repeats. “This was a surprise,” he said. To corroborate, researchers in his lab used CRISPR to insert intermediate-sized repeats into the C9ORF72 gene in isogenic human neuronal precursor cells. Here, too, these repeats boosted expression of isoform 3.
How would this overexpression cause CBD? Lee does not suspect foci of expanded C9ORF72 mRNA found in FTD/ALS, or poly-dipeptides transcribed from said RNA. Instead, he thinks the intermediate repeats cause tau to aggregate. Using tau biosensor cells pioneered by Marc Diamond, Lee found that seeds from CBD brain tissue precipitated new tau aggregates twice as fast when C9ORF72 was overexpressed.
Others were intrigued. Rademakers has seen expansions affect expression in the same way. She asked how that could relate to CBD, or why it would be specific for CBD. Had Lee looked at expansion sizes in progressive supranuclear palsy? Lee said not yet.
Others wondered about expansions of 30 to100 repeats and about growth of expansions with age, since there is evidence that this happens in somatic cells. Lee said he has no expansions of this size in his sample set, but knows of one person carrying 40 to 50 repeats who was clinically normal but had evidence of C9 RNA foci and dipeptide repeats. At best, Lee thinks such repeat sizes might be a risk factor for CBD. He has found no evidence for expansion growth, but said newer, more sensitive techniques would be worth testing.
Multiple Gene Variants
One of the most puzzling hurdles to understanding FTD is that many of the implicated genes lead to extremely heterogeneous clinical presentations. C9ORF72 expansions are found in patients with FTD, ALS, and even psychosis. What causes such pleiotropy? Mathieu Barbier, Brain and Spine Institute, Paris, wondered if it’s other genetic variants. To get at this, he examined age at onset (AAO) among C9 expansion carriers. While their mean AAO was 60, symptoms can strike as early as 30 and as late as 90.
Barbier looked within families to identify other variants that influence onset. Among 333 affected members of 133 families collected through the the French Reference Centre for Rare and Early Dementias, he found parent-offspring and sibling-sibling AAO correlations that suggested a contribution from a variant on the X chromosome. Taking a different tack, he sorted 75 patients from 34 families into those whose AAO was very similar (within two years) or very different (20-plus years apart). Then, using linkage analysis of 20 concordant and 30 discordant pairs of relatives, he found two suggested links to AAO, one again on chromosome X, and another on chromosome 9. On the latter, the closest gene was PTPRD, which encodes a protein phosphatase found in excitatory presynapses. Lo and behold, the closest gene to the X chromosome locus was SLITRK2, which encodes a postsynaptic protein that turns out to interact with PTPRD in mice.
To further narrow down the loci, Barbier genotyped the concordant and discordant pairs. This turned up 12 SNPs in eight loci, including those near SLITRK2 and three other genes involved in synaptic function—CTNNA2, LRRTM1, and DAAM1. As expected, cross-mapping the linkage and association data revealed one SNP, rs1009776, which lies just upstream of the SLITRK2 gene. This polymorphism associated with age of onset, but only in men. In replicating the analysis in a cohort of 124 unrelated C9ORF72 patients from the International Frontotemporal Dementia Genomics Consortium coordinated by Raffaele Ferrari, University College London, only rs1009776 reached significance and again only in men. Barbier found no association between this SNP and risk for ALS or FTD in people who carry GRN mutations, suggesting the association is specific for C9ORF72 disease. He said work is in progress to understand the functional effect of this variant.
As for GRN mutation carriers, Rademakers is part of a worldwide consortium to study the effect of genetic variants on disease risk and age of onset in this population. To date they have genotyped whole genomes of 382 unrelated symptomatic patients who share 120 different GRN mutations among them. The genotyping revealed no variants that affect AAO, but did turn up variants in two loci that modify risk. These were near the GFRA2 and TMEM106B genes.
Previously, Rademakers and colleagues reported that minor alleles in the TMEM106B gene protect C9ORF72 mutation carriers from disease (van Blitterswijk et al., 2014). In Sydney, she said the gene also protects GRN mutation carriers. “TMEM106B has turned into my favorite gene,” quipped Rademakers, believing it holds the key to a natural cure for GRN mutations. Christian Haass, DZNE, Munich, agreed. “This is super important,” he said. “If we can figure out how this gene protects, then we should be able to treat the disease,” he said.
Based on her data, Rademakers said that a GRN mutation carrier who has one copy of the protective TMEM106B allele is half as likely to get sick while most people with two protective variants never get it. She calculated that 14 percent of the general population is homozygous for the protective TMEM106B variant. “Why do we never see GRN mutation carriers with two copies of this TMEM106B allele in the clinic?” she asked. “Because they rarely get FTD.” She urged the field to debate the merits of genetic testing for TMEM106B in GRN carriers. But in a twist, she also showed how complex the interplay can be between genetic variants. She described one GRN mutation carrier who was homozygous for the protective TMEM106B haplotype; this man did not have FTD but Parkinsonism. It turned out he also carried an R816C variant in the CSF1R gene that has been linked to clinical features of PD. “That is likely what caused the Parkinsonism, not the GRN mutation,” she said.—Tom Fagan
References
News Citations
- FTD Gene Hunt Turns Up CYLD and Modifying Factors
- Genetics Tie ALS into the Frontotemporal Dementia Spectrum
- ALS—A Polyglutamine Disease? Mid-length Repeats Boost Risk
Paper Citations
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006 Aug 24;442(7105):920-4. PubMed.
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006 Aug 24;442(7105):916-9. PubMed.
- Rademakers R, Eriksen JL, Baker M, Robinson T, Ahmed Z, Lincoln SJ, Finch N, Rutherford NJ, Crook RJ, Josephs KA, Boeve BF, Knopman DS, Petersen RC, Parisi JE, Caselli RJ, Wszolek ZK, Uitti RJ, Feldman H, Hutton ML, Mackenzie IR, Graff-Radford NR, Dickson DW. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet. 2008 Dec 1;17(23):3631-42. PubMed.
- van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A, Lemmens R, Schelhaas HJ, Groen EJ, Huisman MH, van der Kooi AJ, de Visser M, Dahlberg C, Estrada K, Rivadeneira F, Hofman A, Zwarts MJ, van Doormaal PT, Rujescu D, Strengman E, Giegling I, Muglia P, Tomik B, Slowik A, Uitterlinden AG, Hendrich C, Waibel S, Meyer T, Ludolph AC, Glass JD, Purcell S, Cichon S, Nöthen MM, Wichmann HE, Schreiber S, Vermeulen SH, Kiemeney LA, Wokke JH, Cronin S, McLaughlin RL, Hardiman O, Fumoto K, Pasterkamp RJ, Meininger V, Melki J, Leigh PN, Shaw CE, Landers JE, Al-Chalabi A, Brown RH Jr, Robberecht W, Andersen PM, Ophoff RA, van den Berg LH. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009 Oct;41(10):1083-7. Epub 2009 Sep 6 PubMed.
- Stogmann E, El Tawil S, Wagenstaller J, Gaber A, Edris S, Abdelhady A, Assem-Hilger E, Leutmezer F, Bonelli S, Baumgartner C, Zimprich F, Strom TM, Zimprich A. A novel mutation in the MFSD8 gene in late infantile neuronal ceroid lipofuscinosis. Neurogenetics. 2009 Feb;10(1):73-7. Epub 2008 Oct 11 PubMed.
- Geier EG, Bourdenx M, Storm NJ, Cochran JN, Sirkis DW, Hwang JH, Bonham LW, Ramos EM, Diaz A, Van Berlo V, Dokuru D, Nana AL, Karydas A, Balestra ME, Huang Y, Russo SP, Spina S, Grinberg LT, Seeley WW, Myers RM, Miller BL, Coppola G, Lee SE, Cuervo AM, Yokoyama JS. Rare variants in the neuronal ceroid lipofuscinosis gene MFSD8 are candidate risk factors for frontotemporal dementia. Acta Neuropathol. 2019 Jan;137(1):71-88. Epub 2018 Oct 31 PubMed.
- Nuytemans K, Bademci G, Kohli MM, Beecham GW, Wang L, Young JI, Nahab F, Martin ER, Gilbert JR, Benatar M, Haines JL, Scott WK, Züchner S, Pericak-Vance MA, Vance JM. C9ORF72 Intermediate Repeat Copies Are a Significant Risk Factor for Parkinson Disease. Ann Hum Genet. 2013 Jul 12; PubMed.
- Nuytemans K, Inchausti V, Beecham GW, Wang L, Dickson DW, Trojanowski JQ, Lee VM, Mash DC, Frosch MP, Foroud TM, Honig LS, Montine TJ, Dawson TM, Martin ER, Scott WK, Vance JM. Absence of C9ORF72 expanded or intermediate repeats in autopsy-confirmed Parkinson's disease. Mov Disord. 2014 May;29(6):827-30. Epub 2014 Feb 26 PubMed.
- van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Murray ME, Hsiung GY, Stewart H, Karydas AM, Finger E, Kertesz A, Bigio EH, Weintraub S, Mesulam M, Hatanpaa KJ, White CL 3rd, Strong MJ, Beach TG, Wszolek ZK, Lippa C, Caselli R, Petrucelli L, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Mackenzie IR, Seeley WW, Grinberg LT, Miller BL, Boylan KB, Graff-Radford NR, Boeve BF, Dickson DW, Rademakers R. TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol. 2014 Mar;127(3):397-406. Epub 2014 Jan 3 PubMed.
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.