Are Protein Strains The Cause of Different Tauopathies?
Quick Links
One facet of neurodegeneration that continues to bewilder researchers is that the same miscreant protein can turn up in many different disorders. Take tau, for example. This protein misfolds and aggregates in more than 15 tauopathies, each with its own unique pattern of neurodegeneration and clinical symptoms. Rather than being merely a marker of neurodegeneration, as is widely accepted, could tau seed these disparate pathologies? According to work presented at the Alzheimer's Association International Conference held July 13-18 in Boston, Massachusetts, the answer may well be yes. Marc Diamond, from Washington University, St. Louis, Missouri, reported that tau forms a multitude of strains, each with its own morphology and toxic properties. The researchers have isolated some of these strains from patient brain and passaged them in cells. The findings, which some AAIC attendees considered to be a highlight of the meeting, lend solid support to the idea that different strains of the same protein may underlie different tauopathies. If true, this would have important clinical implications, say researchers.
"These findings tell us it will be possible to describe diseases based on an ensemble of conformers," said Diamond. "That brings a level of specificity and understanding that could greatly impact diagnosis and therapy." The work also indicates that tau forms bona fide prions, according to Diamond, who does not use that term lightly.
Researchers in the field of neurodegeneration have grappled with the term prion. They generally reserve it for disorders that are infectious, including bovine spongiform encephalopathy in cows, and scrapie in sheep. Aβ, tau, α-synuclein and other aggregation-prone proteins that undergo template-directed misfolding are often loosely referred to as prion-like. However, in Boston, Diamond claimed that tau satisfied the basic cell biology properties of true prions: It forms strains that are stably passed from mother cell to daughter cell; it can be taken out of the cell and reintroduced into a naive cell where it continues to propagate; and it forms multiple conformers, or strains that do all of the above. These are all characteristics of yeast prions. "We have demonstrated stable, faithful, indefinite reproduction of tau conformers in living systems," said Diamond, adding that this has never been done before with other than traditional prions.
Scientist who attended the symposium were excited by the findings. "I think this work is just fabulous," said Lary Walker, Emory University, Atlanta, Georgia. "The idea that they can take the aggregates of one morpho-type and propagate it for generations is remarkable."
Tau Strains in Cell Culture
En route to discovering tau prions in diseased human brain, David Sanders, a post-doc in Diamond’s lab, found them in stable cell lines. He used tau fibrils made in vitro to seed aggregation of a tau fragment expressed in monoclonal cells as a yellow fluorescent protein chimera. Sanders chose to express a repeat domain (amino acids 244-372), because full-length tau suppresses cell division and he wanted to propagate the cells for as long as possible. Seeding induced aggregation of the yellow chimera. As the cells grew and multiplied over the next 20 days, Sanders saw a gradual loss of aggregates. However, a small percentage of cells retained obvious yellow inclusions even up to six months and many cell generations later, when the initial tau seed was long gone from the culture medium.
While re-plating cells that propagated these tau aggregates, the researchers noticed something intriguing—as individual colonies grew, they did not all look the same. Some had diffuse perinuclear deposits, others had a few dense inclusions away from the nucleus (see figure below).
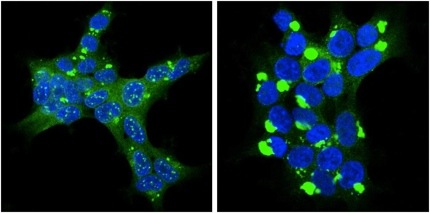
Protein Strains: Tau (green) strains clone 9 (left) and clone 10 have different morphologies. Image courtesy Marc Diamond and David Sanders
In Boston, Diamond reported that these different tau aggregates had different physical properties. On protease digestion, some completely broke apart, while others were resistant, revealing one or two bands on Western blots. The strains seem to have different toxicities, too, said Diamond in response to questions from the audience. Cells poorly tolerate one strain, dubbed clone 9. Cells carrying clone 9 quickly die if stressed, but harbor clone 10 without duress. When taken out of the cell and injected into fresh cells that had never seen tau aggregation, these strains self-propagated through daughter cells, forming identical new inclusions and retaining their characteristic biochemical properties, just like prions do.
Strains from Human Brain
Fascinating, but this could be just an in-vitro phenomenon. To test whether these strains are related to pathological tau aggregates found in the human brain, Sanders seeded aggregation using brain tissue samples taken from people who had had Alzheimer's disease, progressive supranuclear palsy, corticobasal syndrome, Pick's disease, or other tauopathies. At AAIC, Diamond showed that these samples seeded tau aggregates with different morphologies. The inclusions varied from ordered to disordered, from speckled to mosaic, and each seemed to have different propensity to cause cell death, said Diamond. Samples from AD brain tissue all generated cell colonies with speckled inclusions (see image below), except for samples from one patient.
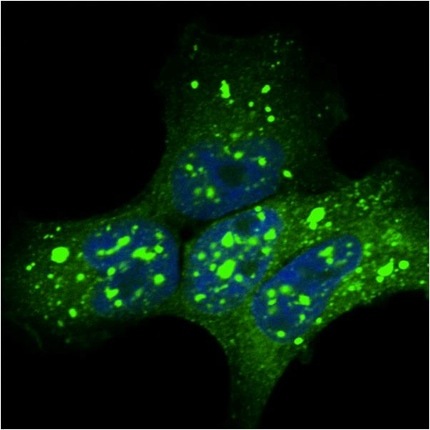
An Alzheimer’s disease tau strain: Tissue samples from AD brain propagated tau strains (green) in monoclonal cells Image courtesy Marc Diamond and David Sanders
Diamond's talk generated a fusillade of audience questions. What is the nature of the speckles? Do the infected cells survive because they have developed compensatory mechanisms? Does one strain dominate over the other? On the latter, Diamond said that clone 9 dominates over clone 10, but he has yet to to characterize the inclusions with biophysical techniques. Diamond is also studying RNA expression in the cells to see if different tau strains elicit different responses.
The findings suggest that the established concept of explaining different tauopathies by the location in the brain where the pathology first emerges may no longer be adequate, Diamond told Alzforum. He believes that location and strain are both important characteristics, as is the mechanism by which tau strains infect cells.
One consequence of this work could be that doctors better characterize, diagnose, and treat tauopathies. Diamond envisages being able to test human tissue samples, perhaps cerebrospinal fluid, or even blood, for tau strains. "We are working on a way to detect these strains by conformation," he told Alzforum. He believes that if he can correlate disease with a particular mix of strains, this could replace standard neuropathological diagnosis and also help development of treatments. Over the long term, Diamond sees therapy, particularly with antibodies, as being strain-specific. In the short term, he said strain-based diagnosis would be a huge help in designing and interpreting clinical trials, which have so far yielded no new drugs for AD or other tauopathies.—Tom Fagan.
References
No Available References
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
ICM
University Hospital Basel
MRC Laboratory of Molecular Biology
This report raises the question whether strains made of four tau repeats are related to pathological tau aggregates found in human brain. The question is answered in the affirmative, because seeds prepared from different human tauopathies gave rise to tau inclusions with different morphologies in cell culture, as judged by their light microscopic appearance.
We would like to note, however, that the report does not mention our published work (Clavaguera et al, 2013), which has provided direct evidence for the existence of distinct tau strains in human brain. We injected brain extracts from humans who had died with Alzheimer’s disease, tangle-only dementia, progressive supranuclear palsy, corticobasal degeneration, argyrophilic grain disease or Pick’s disease into the hippocampus and cerebral cortex of mice transgenic for human wild-type four-repeat tau (line ALZ17). These mice do not form silver-positive tau inclusions or neurodegeneration. Argyrophilic tau inclusions formed in all cases. Also, following the injection of the corresponding brain extracts, we recapitulated the hallmark lesions of the four-repeat human tauopathies, argyrophilic grain disease (silver-positive grains and silver-negative tufted astrocyte-like lesions), progressive supranuclear palsy (silver-positive nerve cell inclusions and silver-positive tufted astrocyte-like lesions) and corticobasal degeneration (silver-positive nerve cell inclusions and silver-positive astrocytic plaque-like lesions).
Similar inclusions also formed after the intracerebral injection of brain homogenates from human tauopathies into non-transgenic mice. In addition, we demonstrated the propagation of tau inclusion formation following the serial injection of different strains into mouse brain.
These findings indicate that distinct tau strains exist, which result in the preferential aggregation of tau in certain cells and brain regions. Once small numbers of tau inclusions have formed in the brain, they may become self-propagating and spread in a prion-like manner, independently of other pathogenic mechanisms.
References:
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M, Ghetti B, Goedert M, Tolnay M. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013 Jun 4;110(23):9535-40. PubMed.
Make a Comment
To make a comment you must login or register.