Brain Conference Spotlights Transcriptomics, Therapeutic Strategies
Quick Links
About 140 scientists gathered in Rungstedgaard, north of Copenhagen, Denmark, May 5–9 for the biannual Brain Conference organized by the Federation of European Neuroscience Societies and the Lundbeck Foundation. Co-chaired by Beth Stevens and Christian Haass, who shared the 2018 Brain Prize with Bart De Strooper, John Hardy, and Michel Goedert (March 2018 news), this meeting focused on understanding and targeting Alzheimer’s disease.
The gathering afforded students, postdocs, and principal investigators opportunities to mingle, discuss the state of the field, and find common ground for future work. It also reflected a changing of the guard in AD research. Sprinkled among reviews of established themes, including the amyloid hypothesis, tau, proteopathic spread, and network dysfunction, were snippets of new information on fast-moving areas, mostly TREM2 and the role of glia in neurodegeneration. “The meeting covered diverse topics and brought together senior scientists who had never met each other before,” noted Haass. “It was a fantastic opportunity to forge new collaborations,” he said.
One such initiative, between Haass, from Ludwig Maximilians University, Munich, and Ido Amit, Weizmann Institute of Science, Rehovot, Israel, will examine an antibody Haass developed to prevent shedding of the extracellular domain of TREM2. His team found that stifling this process by blocking the ADAM10 secretase, which cleaves TREM2, improves microglial phagocytosis, and might point to a therapeutic strategy to treat Alzheimer’s and other diseases. But since ADAM10 cleaves many substrates, including APP, the scientists searched for a more specific way to prevent TREM2 shedding.
Luckily, sheddases such as ADAM10 cut a single peptide bond in the microglial receptor, between histidine 157 and serine 158 in the stalk that separates its transmembrane and immunoglobulin-like type V domains (see image below and Aug 2017 news). Haass reckoned that an antibody that prevents ADAM10 from accessing this region might hinder proteolysis and prevent sTREM2 shedding.
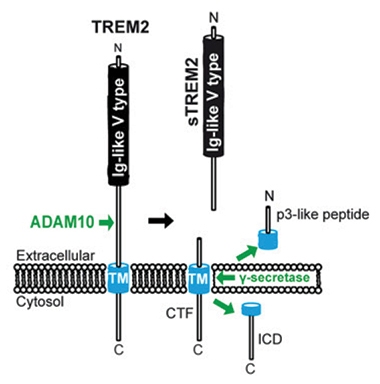
Anti-Shedding. ADAM10 cuts the stalk region in the extracellular domain of TREM2, releasing sTREM2. The monoclonal antibody 4D9 binds to the stalk region, prevents shedding, and boosts microglial function. [Courtesy of Schlepckow et al., EMBO Mol Med 2017.]
This seems to be the case. After using the TREM2 ectodomain as an antigen, the scientists found that one monoclonal antibody, 4D9, reduced release of sTREM2 and enhanced levels of the full-length protein on the cell surface. The 4D9 bound close to histidine 157, elbowing aside ADAM10 and ADAM17’s attempts to bind there. The monoclonal boosted signaling through the TREM2 binding partner DAP12, bolstering macrophages’ ability to survive after their tropic support had been withdrawn. It also improved microglial phagocytosis of myelin particles.
“This all looks promising,” said Hass, but all the same he cautioned that the therapeutic window might be narrow. This is in part because progranulin knockouts that cause FTD super-activate microglia, suggesting that over-silencing TREM2 might be deleterious (May 2016 news). Still, Haass has patented the antibody and licensed it to Denali Therapeutics for development. In the new collaboration, his and Amit’s lab will investigate if 4D9 might treat obesity, a different disorder that is all the same related to Alzheimer’s.
What in the world does TREM2 have to do with obesity? It turns out that macrophages in fat tissue mimic what microglia do in the brain. Previously, Amit reported that disease-associated microglia (DAM) in the brains of mouse models of amyloidosis expressed a specific transcriptomic signature (Jun 2017 news). Might something similar be going on in other conditions or organs?
At Rungstedgaard, Amit reported that adipose tissue in the abdomen contained a specific subset of lipid-associated macrophages. These LAMs express TREM2, and their transcriptome resembles that of DAM microglia. LAMs proliferate when mice eat a high-fat diet. Knocking out TREM2 blocked expression of the LAM transcriptome. This caused adipocytes to swell in mice on a high-fat diet, raised plasma high- and low-density lipoproteins, cholesterol, and glucose, and led to insulin resistance. The findings suggest that TREM2 may be a potential therapeutic target for metabolic diseases, said Amit. Might 4D9 boost LAM function and render adipocyte more functional, as well, then? That’s what the researchers will test.
Others cautioned that because TREM2 knockouts have not just suppressed LAMs but also suppressed microglia, responses in peripheral fat tissue could reflect changes in motor activity, appetite, or other effects in the brain.
Marco Colonna, Washington University, St. Louis, also collaborates with Amit and has previously identified TREM2 as a lipid sensor that binds HDL and LDL. He said the effect of knocking out TREM2 on LAMs is very robust, but that it will be important to define exactly how this alters metabolism. Colonna further reports that TREM2 signaling drives microglial cell metabolism and that cyclocreatine, a small molecule that can regenerate ATP in cells, rescues microglial activity in TREM2 knockouts (Aug 2017 news).
Haass thinks TREM2 might modulate adipose tissue macrophages in the same fashion. Thus, TREM2 would tie energy and fat metabolism in the brain in with that in the periphery. “That would be exciting and totally new biology,” he said.
Adriano Aguzzi and colleagues at University Hospital, Zurich, are using a different antibody-based strategy. They have begun a large effort to screen patient blood samples for autoantibodies that modify disease in a process Aguzzi calls "reverse personalized medicine." The idea is not to start with a specific disease and look for an immunotherapy, but rather to find auto-antibodies and ask what diseases they might induce or modify. “When we find a hit, we go back and ask why the patient was in the hospital and look for connections between their autoimmunity and disease,” he said. The strategy is reminiscent of the one that identified aducanumab. In that case, researchers took plasma from octogenarians who had no signs of AD, and searched for antibodies that might be protecting them. “Of course we have had many therapeutic trials with antibodies that did not work,” Aguzzi admitted, “but some may still prove useful.” He hopes that because human antibody therapeutics work well in many other diseases, it will eventually be possible to find antibodies for neurodegenerative disorders.
Aguzzi is testing plasma from people of all ages. “It always struck me that people with PrP mutations make the abnormal protein from day one, but can live until they are middle-aged and older. Maybe they have immunity that protects them until then,” he speculated. While such antibodies are rare, given that the immune system has evolved to suppress them, he reasoned that if he tests enough plasma samples for enough antigens he might identify those rare instances. University Hospital Zurich agreed to allow him to screen, with their consent, every patient who comes through the door.
Aguzzi has massively scaled up this approach. He has put a system in place that runs 40,000 assays per night for antibodies to a dozen antigens. When there is a hit, the blood sample is harvested the next day for B cells to generate clones for antibody production. If that’s not possible, the donor is contacted via an institutional registry since Aguzzi’s lab has no access to their personal information. “Patients have been incredibly generous, and we have already had many come back to donate more blood,” he said.
To date, Aguzzi’s lab has screened samples from more than 60,000 people. They found autoantibodies to LAG3, a cell-surface protein proposed to be a binding site for α-synuclein oligomers (Oct 2016 news). That person didn’t have Parkinson’s disease, but did have a different disease. Aguzzi declined to say what, and teased the audience by calling it “extremely exciting.”
He is also finding many antibodies to tau. The older the person, the more tau antibodies he or she tends to have. Those antibodies were extremely specific, with high affinity for tau. “It will be important to figure out if they modify disease in any way,” Aguzzi said.
It is not lost on Aguzzi that his large-scale fishing expedition comes against the backdrop of aducanumab’s and crenezumab’s high-profile Phase 3 failure in symptomatic patients, (Jan 2019 news; Mar 2019 news). These trials, as did other immunotherapies and γ- and β-secretase inhibitor trials before them, have put pressure on the amyloid hypothesis (May 2019 conference news). Still, most researchers at Rungstedgaard consider it premature to dismiss Aβ immunotherapy. Dennis Selkoe, Brigham and Women’s Hospital, Boston, argued that neutralizing soluble Aβ oligomers remains viable, if done early enough, and showed data suggesting that aducanumab, bapineuzumab, gantenerumab, and most other anti-Aβ antibodies tested reduced the neurotoxicity of Aβ dimers and small oligomers. Despite reducing these species in the cerebrospinal fluid of 98 patients in the ABBY and BLAZE trials, crenezumab did nothing to slow disease at the mild symptomatic stage (Jul 2018 news). The question, said De Strooper, Dementia Research Institute, U.K., is how early is early enough?
In his talk, De Strooper reviewed the concept that Aβ may be an initial trigger that starts off a pathogenic cascade that then becomes independent of Aβ, rather than a driver that continues to fuel the process (Karran et al., 2011). In the latter scenario, removing some of the peptide should be beneficial even late in disease. But in the former, targeting Aβ after it has reached a threshold concentration in the brain will be too late. “In this worst-case scenario, we need to have a better concept of what ‘early’ means,” De Strooper said.
There are even indications that toxic Aβ species may change during the course of this long disease. This would also have implications for therapeutic efficacy. Mathias Jucker, University of Tuebingen, reviewed data from his lab on proteopathic spread that indicated that Aβ seeds from mice that have no detectable plaques were much more potent than those from mice with established plaques (Ye et al., 2017). This points to a qualitative difference between Aβ seeds in early and late disease.
Jucker reported that only one of five clinically tested anti-Aβ antibodies bound those early seeds. He agreed that immunotherapy may still be a viable strategy, but only if it targets the right Aβ molecules. What those seeds are remains to be proven. They may be structurally different from the dimers and small oligomers that Selkoe’s lab was able to neutralize with the same antibodies.
Transforming Science with Single-Cell Transcriptomics
A plethora of talks and posters at Rungstedgaard focused on single-cell sequencing, a rapidly growing trend in the field. Much of that work is relying heavily on single nuclei, aka snRNA-Seq, to avoid having to isolate whole cells from human postmortem brain tissue. Part of this new work is the TSNE plots (pronounced “tisney” and standing for T-distributed stochastic neighbor embedding) that look like aerial photos of island archipelagos. TSNE plots identify cellular changes in various disease models, showing clusters of cells with similar transcriptomes. This, in turn, allows scientists to easily distinguish any cell type, whether glia, neurons, immune or other cells, and even home in on subpopulations undergoing transcriptomic changes.
In part, single-cell transcriptomics is how scientists are tackling the “cellular phase” of AD as proposed by De Strooper (March 2016 webinar). These types of data, especially once obtained in situ, may allow scientists to parse how individual cells respond to the initial proteopathic triggers of AD long before neurodegeneration and dementia set in.

Transformative Plots. “Tisney” displays transcriptomes of individual cells in two dimensions. The closer the dots, the more similar their transcriptomes. Clusters map to cell types and subtypes. [Courtesy of Keren-Shaul et al., Cell 2017.]
Amit used the technique to identify the LAMs. Colonna used it to identify a population of oligodendrocytes that are activated in the 5xFAD model of AD independently of TREM2 signaling. These cells massively upregulate expression of Serpina3n, an inflammatory protease inhibitor that might prevent clearance of Aβ, Colonna speculated. While recent transcriptomic analysis of the frontal cortices of AD patients suggests there are fewer oligodendrocytes in disease (May 2019 news), Colonna said his mouse data suggest a more functional change in oligodendrocytes. “We see less myelination and more pro-inflammatory signals from these cells,” he said.
Besides studying how oligodendrocytes change in AD, Colonna used snRNA-Seq to compare transcription profiles of disease-associated microglia from 5xFAD mice and microglia from tissue samples of 50 AD patients, including 10 with the TREM2 R47H mutation and 10 with the R62H mutation. In Rungstedgaard, he reported that the human and mouse signatures only partially overlapped. “That’s not surprising, since the mouse is principally a model of amyloidosis and lacks tau pathology,” Colonna said. He also saw that microglial gene expression was generally lower in brain tissue from the TREM2 mutation carriers, which is in keeping with TREM2 being a driver of disease-associated microglial transcriptomes.
The conference also showed, however, that scientists are still debating how to interpret TSNE plots. For example, since the data comes from nuclei isolated from postmortem brain, it says little about the specific anatomical origin of the cells—an important omission for a tissue as supremely patterned as the brain. In her talk, Stevens, Boston Children’s Hospital, noted that one microglial cluster in early postnatal development might have gone unnoticed had she not seen that it was specifically near axon tracts. It turned out to play a major role in synaptic pruning.
A better understanding of transcriptomic reprogramming may come from in situ hybridization to localize these changes, from in situ single-nucleus RNA-Seq, which is in its infancy, or from techniques such as spatial transcriptomics, as described by Ashley Lu from De Strooper’s lab at KU Leuven, Belgium. She profiled gene expression in tiny blocks of tissue, at 500 brain locations, and across four different time points, in APP knock-in mice (APPNL-G-F).
Lu’s goal was to compare transcriptomes at different distances from plaques. She identified a cluster of 57 co-expressed genes that are upregulated in close proximity to the amyloid deposits. These plaque-associated genes are both micro- and astroglial, and are known players in neuroinflammation, suggesting that plaque directly triggers this response. The findings also suggest cellular cross-talk in the vicinity of plaques, she concluded.
While new transcriptomics techniques are generating reams of data, scientists admit that they still don’t know exactly what it all means. “Ultimately, we need to figure out how all these transcriptomic structures relate to function,” Stevens emphasized.—Tom Fagan
References
News Citations
- De Strooper, Goedert, Hardy, Haass Share 2018 Brain Prize
- TREM2 Cleavage Site Pinpointed: A Gateway to New Therapies?
- Microglia Prune Synapses in a Subtype of Frontotemporal Dementia
- Hot DAM: Specific Microglia Engulf Plaques
- Without TREM2, Microglia Run Out of Gas
- Immune Receptor May Smuggle α-Synuclein into Neurons, Hasten Proteopathy
- Roche Pulls Plug on Two Phase 3 Trials of Crenezumab
- Biogen/Eisai Halt Phase 3 Aducanumab Trials
- Keep Your Enthusiasm? Scientists Process Brutal Trial Data
- On Target: Crenezumab Reduces Aβ Oligomers in CSF
- When It Comes to Alzheimer’s Disease, Do Human Microglia Even Give a DAM?
Therapeutics Citations
Webinar Citations
Research Models Citations
Paper Citations
- Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011 Sep;10(9):698-712. PubMed.
- Ye L, Rasmussen J, Kaeser SA, Marzesco AM, Obermüller U, Mahler J, Schelle J, Odenthal J, Krüger C, Fritschi SK, Walker LC, Staufenbiel M, Baumann F, Jucker M. Aβ seeding potency peaks in the early stages of cerebral β-amyloidosis. EMBO Rep. 2017 Sep;18(9):1536-1544. Epub 2017 Jul 12 PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.