Could Benefit of Plaque Removal Grow in Time?
Quick Links
Consensus is strengthening among Alzheimer’s researchers that antibodies that abolish plaque nudge down the rate of cognitive decline. Alas, the effect is so small—is it meaningful? At the Alzheimer’s Association International Conference, held July 31-August 4 virtually and in San Diego, California, several presenters quantified the clinical benefit of amyloid removal and attempted to forecast the effects of long-term treatment. They admitted that the cognitive benefit in trials has been tiny, but drug sponsors argued that the difference between treatment and placebo groups is likely to grow over time.
- As a class, plaque-clearing anti-amyloid antibodies slow cognitive decline.
- The benefit is minute. Progression models predict it will get bigger in time.
- As a result, treatment could delay progression to dementia.
At Roche, Norman Mazer developed the “Quantitative ATN” (Q-ATN) model of AD to predict changes in cognition as a function of amyloid removal. He used data from numerous observational studies to derive mathematical relationships. The model’s predictions correlated well with actual clinical trial results. A simulated five-year course of the company’s anti-amyloid antibody gantenerumab forecast that the CDR-SB difference between the treatment and placebo groups would expand as the disease progression curve flattens. Evaluating a different metric, researchers at Eisai estimated that long-term treatment with their drug, lecanemab, could delay progression from mild cognitive impairment to dementia by more than three years. Upcoming trial results from both antibodies expected this fall will provide a reality check to these company models, and help refine their predictions.
Rachelle Doody at Roche believes the Q-ATN model could affect how such trial results are interpreted. “The clinical significance of the CDR-SB score at endpoint should be evaluated in the context of accrued benefits over time, especially once this model is prospectively validated with longitudinal data,” she wrote to Alzforum.
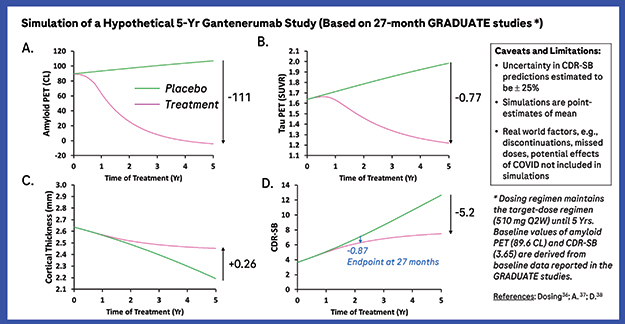
Stanch Progression? Roche’s mathematical Q-ATN model foresees a drop in plaque (upper left), followed closely by a drop in the tau PET signal (upper right). After a delay, the brain shrinkage slows down (lower left), stabilizing cognition (lower right). [Courtesy of Roche.]
Is the Cognitive Benefit Real?
Trials of early immunotherapies, such as bapineuzumab and solanezumab showed little effect on either plaques or cognition (Jan 2014 news; Dec 2016 conference news). Ditto for initial trials of later antibodies, such as the SCarlet RoAD and Marguerite RoAD studies of low-dose gantenerumab (Dec 2014 news; Nov 2015 conference news). In subsequent studies, however, high doses of four antibodies—aducanumab, gantenerumab, lecanemab, and donanemab—proved able to banish plaques, while lightly tapping the brakes on cognitive decline, by 20 to 40 percent (Aug 2018 conference news; Dec 2019 conference news).
At AAIC, Nicolas Villain of Sorbonne University in Paris, presented a meta-analysis of only the high-dose arms from the lecanemab and donanemab Phase 2 trials and the two aducanumab Phase 3 trials, EMERGE and ENGAGE. In the combined analysis, the drugs had a statistically significant cognitive benefit after 18 months, slowing decline on the CDR-SB by an average of 0.24 points, and on the ADAS-Cog by 1.25 points. Villain’s result contrasts with published meta-analyses of diverse amyloid-targeting therapies, many having little effect on plaque. Those analyses found no consistent cognitive benefit (Ackley et al., 2021; Richard et al., 2021). Villain said the size of the effects on both endpoints he looked at were far below what is considered the minimal clinically relevant difference, i.e., 1.63 points for the CDR-SB and 3.8 for the ADAS-Cog. In addition, about one in 200 participants in these trials developed serious symptomatic brain edema or microhemorrhages (ARIA). Thus, the risk/benefit ratio of short-term anti-amyloid immunotherapy is high, Villain concluded.
Other analyses also concluded that the cognitive benefit of amyloid removal is real. Yaning Wang of Createrna Science and Technology, Clarksburg, Maryland, a spin-off of the Chinese biotech firm Wuhan QR Pharma, previously worked at the Food and Drug Administration. He was part of the team that evaluated aducanumab’s licensing application. At AAIC, Yang defended the agency’s decision to grant accelerated approval despite a negative AdComs evaluation. After the FDA advisors had pointed out that there was no way to know whether the positive EMERGE or the negative ENGAGE aducanumab trial was the anomaly, FDA scientists analyzed available clinical trial results from all antibodies in this class. At the time, published data consisted of aducanumab, gantenerumab, lecanemab, crenezumab, bapineuzumab, and solanezumab trials. Wang said that the FDA found a consistent linear relationship between amyloid removal and cognitive benefit across these antibody trials, even for negative ones. ENGAGE was the only trial that did not fall on this line, leaving the FDA to conclude that it was the outlier.
Wang also talked about effect size. He noted that because cognition declines slowly at the prodromal stage of AD, the tiny, measured benefit on the CDR-SB in EMERGE represented a 25 percent slowing. To achieve the minimum clinically relevant difference on CDR-SB in 18 months, the drug would have had to completely halt progression, an unrealistic standard, Wang said.
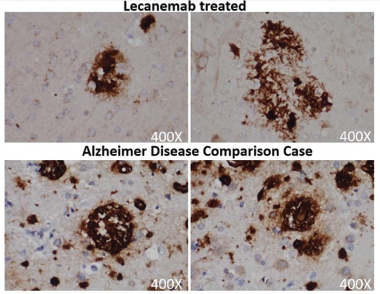
Chewed Up Plaques? Amyloid deposits (brown) in a person treated with lecanemab (top) have a “moth-eaten” appearance compared to the denser plaques in an untreated AD patient (bottom). [Courtesy of Honig et al.]
Data at AAIC also helped lay to rest the niggling question of whether anti-amyloid antibodies truly clear plaque, or instead lower the amyloid PET signal in some other way, such as coating amyloid deposits and preventing tracer binding. This concern has not yet been definitively settled, since few people who have had high-dose anti-amyloid immunotherapy have passed away and had an autopsy. In San Diego, Lawrence Honig of Columbia University Irving Medical Center in New York City presented one such case. This man with mild cognitive impairment had received 10 mg/kg lecanemab monthly for 18 months as part of the Phase 2 trial, which reduced his amyloid PET signal from 45 to 26 centiloids. After a two-year gap, he joined the open-label extension and received 10 mg/kg biweekly for two more years, though Honig did not show PET scans from the OLE. The man died of an unrelated heart condition at the age of 85.
His autopsy showed that his brain contained almost no diffuse plaques, and only a few loose neuritic plaques that had a “moth-eaten” appearance. His brain had widespread neurofibrillary tangles of tau; likewise, these were not dense as they typically are, but had a thread-like consistency. The man’s brain was assessed as Thal Phase 2, indicating little amyloid, and Braak stage 6, indicating advanced tangle pathology. This combination is rare, making up only 2 percent of cases in the National Alzheimer’s Coordinating Center brain bank, Honig noted.
The results strengthen the case that anti-amyloid immunotherapy truly removes plaque. They dovetail with a previous report in the literature, of a woman who received 32 doses of aducanumab. At autopsy, she had little plaque and widespread tangles of low density (for paper and expert commentary, see Plowey et al., 2022).
Will It Grow?
Do benefits of amyloid removal build up over time, as might be expected for a disease-modifying therapy? To address this, Mazer and colleagues at Roche modeled the amyloid-cascade hypothesis. In its simplest form, it posits that plaques unleash tau aggregation, which in turn accelerates brain atrophy, degrading cognition. Mazer expressed these relationships mathematically, deriving equations that described the observed biomarker changes at each step. Natural history studies from several sources, including unpublished imaging data from the Harvard Aging Brain Study, tied plaques to excess tau production (Johnson et al., 2020). From data in multiple publications, the authors found a straightforward relationship between tangle accumulation in the medial temporal cortex and the rate of cortical thinning in that area. The link between thickness in the medial temporal cortex and the CDR-SB came from a study by Brad Dickerson at Massachusetts General Hospital, Boston (Dickerson et al., 2009).
Combining these equations allowed the authors to project how amyloid accumulation might affect the CDR-SB score. They tested their model on additional natural history datasets, and found that the simulated change in biomarkers and CDR-SB closely matched actual findings, suggesting the equations described real biological relationships (Delor et al., 2013; Kim et al., 2020).
But can this model predict the effects of amyloid removal? The researchers ran simulations of how amyloid clearance by aducanumab would affect tangle pathology and CDR-SB, and compared those to actual data from EMERGE and the tau PET substudy. The results agreed well, Mazer claimed. Extending the analysis to published trials of gantenerumab, lecanemab, donanemab, and bapineuzumab, the Q-ATN model accurately predicted CDR-SB change in five of the seven trials, with the discrepancies being the ENGAGE high-dose arm and the SCarlet RoAD trial. “The model is able to explain the lowering of tau PET,” Mazer told Alzforum. “As you remove amyloid, you dial down the inherent drive to generate tau tangles.”
Finally, the researchers used Q-ATN to forecast the results of five years of treatment with gantenerumab. The model predicts that plaques would recede over the first two years, sinking below the positivity threshold before tailing off over the next three years to reach zero. Tau PET would mirror this, but with a more gradual time course and a smaller overall reduction. The rate of cortical shrinkage would start to slow around two years, preserving the remaining thickness of the medial temporal lobe. CDR-SB decline would level off around the same time, reaching statistical difference from placebo of 0.87 points at 27 months, the stopping point for the Phase 3 GRADUATE trials of gantenerumab. The model projects that this difference would expand to 5.2 points after five years, as the placebo group continues to decline faster than the treatment group (see image above).
“Because the rate of cortical atrophy decreases, it bends the curve and leads to the growing separation between the untreated and treated CDR-SB curves,” Mazer said. Crucially, the Q-ATN model suggests that cognitive benefits will be delayed relative to amyloid removal. “There is an inherent lag in the system, because tau influences the rate of change of the cortical thickness. It takes a year or two to see a difference in thickness,” Mazer explained. The data have been submitted for publication.
Is It Meaningful?
How would such a subtle effect—slightly slower decline on cognitive tests—actually improve people’s lives? Amir Tahami Monfared and colleagues at Eisai addressed this using the AD Archimedes condition-event (ACE) simulator developed by the contract research organization Evidera (Kansal et al., 2018). Like the Q-ATN model, AD ACE simulates clinical progression based on biomarker changes; its equations were derived from ADNI observational data. Rather than focus on cognitive scores, AD ACE predicts when symptoms will worsen to the next stage of disease.
Eisai researchers selected 429 of the 1,735 ADNI participants in the AD ACE simulator database, choosing those who best matched the demographic characteristics of participants in the lecanemab Phase 2 trial. In this trial, treatment slowed progression on the CDR-SB by 26 percent. The matched ADNI cohort, with a mean age of 72 and MMSE of 26, provided a glimpse of how disease would progress in this population without treatment. Taking into account the typical remaining lifespan of AD patients and older adults, Eisai researchers projected that continuing lecanemab treatment until the end of a person’s life would result in 7 percent fewer people progressing to mild dementia, 13 percent fewer to moderate dementia, and 10 percent fewer to severe. Six percent fewer people would be institutionalized.
Looking at the data another way, Eisai researchers estimated that the time to each stage of dementia would be delayed by 2.5 to 3 years. People on lecanemab would remain in the community about one year longer and live one year longer on average, gaining 0.75 “quality-adjusted life years” (QALYs). The modeled benefits were greater in younger participants. In a subset with a mean age of 65, progression to mild and moderate AD was projected as being delayed by 3.3 and 3.4 years, respectively (Tahami Monfared et al., 2022).—Madolyn Bowman Rogers
References
Therapeutics Citations
News Citations
- Phase 3 Trial Data for Solanezumab and Bapineuzumab Released
- CTAD: Solanezumab Seen to Nudge AD Ever so Slightly
- End of the RoAD for Gantenerumab? Roche Declares Prodromal Alzheimer’s Trial Futile
- Gantenerumab, Aducanumab: Bobbing Up and Down While Navigating Currents of Trial Design
- Four Immunotherapies Now Banish Amyloid From the Brain
- Amyloid Clearance: Check. Cognitive Benefit: Um … Maybe.
Paper Citations
- Ackley SF, Zimmerman SC, Brenowitz WD, Tchetgen Tchetgen EJ, Gold AL, Manly JJ, Mayeda ER, Filshtein TJ, Power MC, Elahi FM, Brickman AM, Glymour MM. Effect of reductions in amyloid levels on cognitive change in randomized trials: instrumental variable meta-analysis. BMJ. 2021 Feb 25;372:n156. PubMed.
- Richard E, den Brok MG, van Gool WA. Bayes analysis supports null hypothesis of anti-amyloid beta therapy in Alzheimer's disease. Alzheimers Dement. 2021 Jun;17(6):1051-1055. Epub 2021 May 31 PubMed.
- Plowey ED, Bussiere T, Rajagovindan R, Sebalusky J, Hamann S, von Hehn C, Castrillo-Viguera C, Sandrock A, Budd Haeberlein S, van Dyck CH, Huttner A. Alzheimer disease neuropathology in a patient previously treated with aducanumab. Acta Neuropathol. 2022 Jul;144(1):143-153. Epub 2022 May 17 PubMed.
- Johnson K et al. Critical threshold of elevated amyloid associated with rapid tau accumulation: a ca-tau-strophe in the making. HAI Book 2020/#346. HAI Book 2020.
- Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, Grodstein F, Wright CI, Blacker D, Rosas HD, Sperling RA, Atri A, Growdon JH, Hyman BT, Morris JC, Fischl B, Buckner RL. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex. 2009 Mar;19(3):497-510. PubMed.
- Delor I, Charoin JE, Gieschke R, Retout S, Jacqmin P. Modeling Alzheimer's Disease Progression Using Disease Onset Time and Disease Trajectory Concepts Applied to CDR-SOB Scores From ADNI. CPT Pharmacometrics Syst Pharmacol. 2013;2:e78. PubMed.
- Kim KW, Woo SY, Kim S, Jang H, Kim Y, Cho SH, Kim SE, Kim SJ, Shin BS, Kim HJ, Na DL, Seo SW. Disease progression modeling of Alzheimer's disease according to education level. Sci Rep. 2020 Oct 8;10(1):16808. PubMed.
- Kansal AR, Tafazzoli A, Ishak KJ, Krotneva S. Alzheimer's disease Archimedes condition-event simulator: Development and validation. Alzheimers Dement (N Y). 2018;4:76-88. Epub 2018 Feb 16 PubMed.
- Tahami Monfared AA, Tafazzoli A, Ye W, Chavan A, Zhang Q. Long-Term Health Outcomes of Lecanemab in Patients with Early Alzheimer's Disease Using Simulation Modeling. Neurol Ther. 2022 Jun;11(2):863-880. Epub 2022 Apr 25 PubMed.
Other Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
Daping Hospital
It seems that the current 1.5 year research period is not long enough to observe the benefits of Aβ removal or other disease-modifying therapies for AD. A longer time period should be considered in the design of further trials.
A potential adverse effect is that mobilization of Aβ deposits by the investigational drug would increase the generation of Aβ oligomers, which may cause further damage to neurons (Liu et al., 2015). We call this the "dust-raising" effect (Liu et al., 2012). As in a previous autopsy study, the soluble levels of Aβ were increased in the brains of participants who received anti-Aβ immunotherapies (Patton et al., 2006). The dust-raising effect may explain why the brain atrophy was accelerated after immunotherapy (Fox et al., 2005).
References:
Liu YH, Bu XL, Liang CR, Wang YR, Zhang T, Jiao SS, Zeng F, Yao XQ, Zhou HD, Deng J, Wang YJ. An N-terminal antibody promotes the transformation of amyloid fibrils into oligomers and enhances the neurotoxicity of amyloid-beta: the dust-raising effect. J Neuroinflammation. 2015 Aug 28;12:153. PubMed.
Liu YH, Giunta B, Zhou HD, Tan J, Wang YJ. Immunotherapy for Alzheimer disease-the challenge of adverse effects. Nat Rev Neurol. 2012 Jul 3;8(8):465-9. PubMed.
Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickle GD, Luehrs DC, Kuo YM, Lopez J, Brune D, Ferrer I, Masliah E, Newel AJ, Beach TG, Castaño EM, Roher AE. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer's disease patients: a biochemical analysis. Am J Pathol. 2006 Sep;169(3):1048-63. PubMed.
Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, Koller M. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005 May 10;64(9):1563-72. PubMed.
Sorbonne University - APHP - Pitié-Salpêtrière Hospital
Thank you for citing our work with Dr. Vincent Planche regarding our meta-analysis of high-clearance anti-amyloid immunotherapies. The complete manuscript has just been accepted and will be published soon.
All these simulations of the putative long-term effect of these drugs from the companies are promising and revive hope for our patients. Unfortunately, the recent discrepancy between the PRIME, EMERGE, and ENGAGE trial results also reminds us of the limits of simulations and the need to remain humble and avoid false hope in the field.
The postmortem data are encouraging but should also be put in the context of past results, such as the failure of the AN1792 active immunotherapy that also produced similar postmortem data with evidence of high amyloid clearance, an effect on some aspects of tauopathy, etc. (Holmes et al., 2008; Nicoll et al., 2019; Boche et al. 2010). Nonetheless, the long-term clinical efficacy of AN1792, in the few patients with similar characteristics as those currently included (i.e., who did not evidence meningoencephalitis, had a high MMSE at inclusion, a high titer of serum anti-Aβ antibodies, postmortem evidence of almost complete Aβ plaque removal; e.g., individuals 7 and 8 from Holmes et al., 2008), did not appear to be spectacular: “[They] had severe end stage dementia in the absence of any pre-terminal acute confusional state (MMSE score 0) at their last examination before death [i.e., 60-64 months after the inclusion in the study]; Holmes et al., 2008)”.
In conclusion, it is urgent to await more data before planning the future of these therapies, and to remain aware of the limitations of unvalidated surrogate markers (Planche and Villain, 2021).
References:
Boche D, Denham N, Holmes C, Nicoll JA. Neuropathology after active Abeta42 immunotherapy: implications for Alzheimer's disease pathogenesis. Acta Neuropathol. 2010 Sep;120(3):369-84. PubMed.
Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008 Jul 19;372(9634):216-23. PubMed.
Nicoll JA, Buckland GR, Harrison CH, Page A, Harris S, Love S, Neal JW, Holmes C, Boche D. Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer's disease. Brain. 2019 Jul 1;142(7):2113-2126. PubMed.
Planche V, Villain N. US Food and Drug Administration Approval of Aducanumab-Is Amyloid Load a Valid Surrogate End Point for Alzheimer Disease Clinical Trials?. JAMA Neurol. 2021 Nov 1;78(11):1307-1308. PubMed.
The ongoing debate regarding the predicted clinical efficacy of amyloid-targeted and specifically plaque-removal therapies calls to mind the old quote that “the lady doth protest too much.”
I cannot think of another therapeutic strategy that has endured so many failures, and has produced so little of the anticipated benefit, that continues to enjoy such a dedicated following of believers. While we all must agree that the ability of recent amyloid targeted monoclonal antibodies to clear plaque is impressive, the trivial benefit seen would seem to prove that clearing plaque makes little difference, a point further supported by Larry Honig’s autopsied case of Thal stage 2, Braak stage 6.
I am struck by the competing arguments that such little difference is nonetheless better than nothing and so must be good enough, versus the benefit will continue to snowball over time, though if so it will have to be vastly accelerated over the 20-year lag between initial plaque deposition and the eventual emergence of symptomatic memory decline (MCI).
Neither accounts for the mismatch between the topography of clinical deficits and amyloid distribution (Ossenkoppele et al., 2016), a challenge Hardy and Allsop themselves identified in their original 1991 hypothesis paper (Hardy and Allsop, 1991). Nor do these models address any of the other roles that derivatives of amyloid precursor protein play in brain development, maintenance, and function (e.g., Muller et al., 2012; Hick et al., 2015; Richter et al., 2018; Rice et al., 2019; Haass and Willem, 2019).
While I personally lack the expertise to critique the mathematics of these models, those clinical and biological facts argue that there must be more to the story than the underlying assumption on which they are based of a simple gain of Aβ toxicity.
References:
Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, O'Neil JP, Janabi M, Lazaris A, Cantwell A, Vogel J, Santos M, Miller ZA, Bettcher BM, Vossel KA, Kramer JH, Gorno-Tempini ML, Miller BL, Jagust WJ, Rabinovici GD. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain. 2016 May;139(Pt 5):1551-67. Epub 2016 Mar 8 PubMed.
Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991 Oct;12(10):383-8. PubMed.
Müller UC, Zheng H. Physiological Functions of APP Family Proteins. Cold Spring Harb Perspect Med. 2012 Feb;2(2):a006288. PubMed.
Hick M, Herrmann U, Weyer SW, Mallm JP, Tschäpe JA, Borgers M, Mercken M, Roth FC, Draguhn A, Slomianka L, Wolfer DP, Korte M, Müller UC. Acute function of secreted amyloid precursor protein fragment APPsα in synaptic plasticity. Acta Neuropathol. 2015 Jan;129(1):21-37. Epub 2014 Nov 29 PubMed.
Richter MC, Ludewig S, Winschel A, Abel T, Bold C, Salzburger LR, Klein S, Han K, Weyer SW, Fritz AK, Laube B, Wolfer DP, Buchholz CJ, Korte M, Müller UC. Distinct in vivo roles of secreted APP ectodomain variants APPsα and APPsβ in regulation of spine density, synaptic plasticity, and cognition. EMBO J. 2018 Jun 1;37(11) Epub 2018 Apr 16 PubMed.
Rice HC, de Malmazet D, Schreurs A, Frere S, Van Molle I, Volkov AN, Creemers E, Vertkin I, Nys J, Ranaivoson FM, Comoletti D, Savas JN, Remaut H, Balschun D, Wierda KD, Slutsky I, Farrow K, De Strooper B, de Wit J. Secreted amyloid-β precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science. 2019 Jan 11;363(6423) PubMed.
Haass C, Willem M. Secreted APP Modulates Synaptic Activity: A Novel Target for Therapeutic Intervention?. Neuron. 2019 Feb 20;101(4):557-559. PubMed.
University of Alabama, Birmingham
University of Southern California Keck School of Medicine
Of course, clinical and biomarker effects could grow. If your model is projecting the slope of an existing difference it will grow over time, assuming that the conditions remain constant. That is, if the benefits of the drug in reducing plaque are durable and remain the same; if the mortality in the comparison cohort doesn’t alter the validation of the results; if compliance to the treatment regimen remains constant; if the entrance criteria into the trial are applicable for years to come and new comorbidities and other events do not impact the results.
One of the problems with these simulations is that you get what you program. The Roche- and Eisai-sponsored simulations seem to ignore any chance of treatment making people worse either by side effects, ARIA, or from some imbalance caused by the plaque removal. Small differences ultimately will lead to large differences if we wait long enough, i.e., the definition of nonparallel lines from the same starting point.
Another key simulation assumption is whether the modeling is focused on absolute reductions or percentage reductions. Often the models are conducted using percentage reductions, which may work within the limited range of eligibility. However, over the longer term the percentage reductions may diminish as pathology accumulates or as a symptomatic treatment effect fades. Maintaining a constant percentage reduction requires what’s been called a “disease-modifying treatment effect,” meaning that the absolute treatment effect will become larger over time as the placebo decline accelerates over time.
For instance, the 27-month GRADUATE studies displayed in the figure have an absolute treatment effect of about 0.87 in CDR-SB at month 27, corresponding to a percentage treatment effect about 29 percent (0.87/3; 3 is the estimated placebo decline based on the figure from slightly below 4 to slightly above 6); whereas at year 5, the percentage treatment effect is about 58 percent (5.2/9; 9 is the estimated placebo decline based on the figure). Similarly, removing an absolute amount may not maintain the projected difference, as it may depend on the rate of accumulation versus amount removed.
Another curious aspect is that the simulations appear to not be based on cognitive or clinical trends, but rather on biomarkers that show great change with treatment and imply a continued belief in a direct relationship of the biomarkers to clinical benefit. In the Phase 2 donanemab trial, the initial symptomatic treatment effect faded quickly from week 52 to 76 as the donanemab group declined slightly faster than placebo and the relative treatment effect shrank rapidly from approximately 66 percent to 32 percent (See Figure 2A and 2B in Mintun et al., 2021).
In life, they say grief is the price you pay for the joy you’ve had with someone you’ve lost; modeling early Alzheimer studies may be the same.
References:
Mintun MA, Lo AC, Duggan Evans C, Wessels AM, Ardayfio PA, Andersen SW, Shcherbinin S, Sparks J, Sims JR, Brys M, Apostolova LG, Salloway SP, Skovronsky DM. Donanemab in Early Alzheimer's Disease. N Engl J Med. 2021 May 6;384(18):1691-1704. Epub 2021 Mar 13 PubMed.
Alzheimer's Prevention Initiative, Banner Alzheimer's Institute
"The Quantitative ATN” (Q-ATN) model of AD developed by Roche suggests the possibility that the clinical benefits of the “plaque-busting” antibodies are increasingly evident over time. It highlights the likely disconnect between how long trial sponsors would like disease-modifying clinical trials to be, and how long these trials need to be to demonstrate robust treatment effects.
It should be noted that this model uses a number of unproven assumptions, particularly with respect to the relationship between amyloid deposition and tau spreading, and relies on a limited number of antibodies, some with imperfect data sets, such as either large amounts of missing data (aducanumab) or imbalances between treatment groups (ApoE4 in lacanemab).
Fortunately, several large Phase 3 trials will read out in the near future that can inform the model. Overall, the Roche group should be congratulated on their innovative approach to better understand clinical trial results of disease-modifying treatments in AD.
Mayo Clinic College of Medicine
Two groups, one from Eisai and the other from Roche, both with about-to-read-out phase 3 trial programs, reported at AAIC on the modeling of long-term benefits of anti-amyloid monoclonal antibody (AAMA) therapy. The Eisai model was based on data from their phase 2 trial and natural history data for projecting differences in outcome between treated and untreated persons with MCI or mild dementia due to AD. They predicted an eventual several years-worth of delay in progression to moderate dementia. The modeling exercise presented by Roche on long-term consequences of AAMA use was based on a 4-step sequence of events from amyloidosis to tauopathy to cortical atrophy to cognitive and functional change. The Roche modeling also predicted treatment benefits that grew substantially in the out-years after the first 1.5 years of treatment.
Both approaches depend on shaky assumptions. The Eisai model assumes that the magnitude of the benefit seen in their phase 2 trials will be replicated in their phase 3 trial, and that the benefit accrues broadly regardless of age, sex, initial disease severity, etc. The Roche modelers built into their model the assumption that amyloid lowering invariably mitigates effects on cognitive decline.
Even accepting the conjecture that one or more of the to-be-reported AAMAs will show genuine benefits, those benefits are likely to be small (a la donanemab phase 2). Then, the questions are: Are the benefits of AAMAs persistent and independent of disease stage, or do they attenuate as the disease progresses? Are the clinical benefits correlated with amyloid and tau biomarkers? These questions are at the crux of the AAMA story. Answers to these questions will emerge only when the phase 3 to-be-reported trials announce their results later this year and next year.
There should also be long-term extension data that becomes available from the upcoming AAMA trial result reporting, and one would hope that Biogen will finally make available their open-label long-term extension data that were contiguous with the original trials. The long-term open-label data would provide some unique insight into persistence of benefits. In the face of modest actual clinical benefits at 18 months in double-blind trials, I suspect that the sponsors and other advocates will invoke the Eisai and Roche models to claim long-term benefits. But… the Achilles heels of the models is the lack of hard data on clinical benefits of AAMA’s beyond 18 months to support the models’ assumptions
Pentara Corp
Pentara
Pentara Corporation
The models presented above are interesting, but the projections are tenuous, and require untestable assumptions. Just understanding the potential implications of disease modification should motivate us to collect longer-term data, perhaps in open-label extension studies. It reminds me of a T-shirt I saw at the Joint Statistical Meetings several years ago: "Friends don't let friends extrapolate." But if we take these extrapolations as an illustration of a point, they are very useful. The point is that disease modification is not the same as a symptomatic treatment effect.
For decades we've waited for disease modifying treatments for Alzheimer's disease. We are finally seeing data from clinical trials that are consistent with disease modifying effects. But we are so accustomed to interpreting results in the context of symptomatic effects that it is difficult to properly interpret what we are seeing. Treatments that modify the underlying disease process and have effects that truly build up over time essentially extend the time patients function at a particular level during the treatment administration phase. Interpreting these treatment effects as either absolute point changes, percent changes, or even percent slowing does not align with extending functional time.
Since these treatments are likely to be most effective when first given in the early stages of disease when clinical outcomes are changing slowly (top part of a reverse s-curve), extending the time patients function at a particular level will correspond to a percent slowing that decreases over time even if the acceleration factor (deceleration in this case) is constant. Instead of interpreting whether a difference between treatments is clinically meaningful based on mean differences in points on a scale, the amount of time of extended function should be directly interpreted for clinical meaningfulness. This quantity can be estimated from the scales that are currently used in clinical trials, either individually or preferably combining multiple clinical outcomes. Treatments currently in development are demonstrating effects that are consistent with disease modification. The time savings—that is, the extended time of functioning, even over 18 months of treatment—is clinically meaningful, without needing to extrapolate about what was not observed.
Brown University
This is not a rigorous application of a mathematical model; this is an extrapolation that should be viewed skeptically. The extrapolation depends entirely on an unvalidated biological model based on naturally occurring changes in amyloid. Since amyloid would almost never be observed to decline in observational studies, we do not know if these relationships between changes in amyloid, tau, and cortical thickness would be expected to hold in a context where amyloid decreases due to drug treatment. That is, we are extrapolating beyond our observations in assuming how amyloid reductions affect downstream variables up to five years out. There is currently no quality data to support assumptions regarding the long-term cognitive effects of drug-induced reductions in amyloid: We have only observed short-term effects (around 18 months) of drug-induced changes in amyloid and long-term effects of amyloid increases due to Alzheimer’s disease in observational data.
The purpose of a mathematical model in this context is to determine the logical implications of a specific set of assumptions about the biology and the effects of these drugs on amyloid and other biomarkers on long-term cognitive change. These models are notoriously vulnerable to details of the input assumptions, and so each must be carefully evaluated and validated against established benchmarks. No such details have been provided for the Q-ATN model. Based on limited available information online, the Q-ATN model is used to produce simulated data on time-changing amyloid and tau levels, cortical thickness, and cognition, to compare the long-term difference in cognition for drug-treated and untreated simulated individuals for five years total. Mazer et al. parameterize the effect of amyloid on tau, the effect of tau on cortical thickness, and the effect of cortical thickness on cognition in this model over time using observational data.
We have some indirect indications that benefits of amyloid reduction do not grow with time. First, the EMBARK trial re-enrolled eligible individuals from EMERGE and ENGAGE. After a gap period of 1.1 to 3 years (median 1.8 years) after the initial 78 weeks of treatment, the cognitive difference between treatment and placebo groups did not grow, and, if anything, the two groups started to converge (CTAD 2021). Second, as mentioned in the conference story above and a separately reported autopsy study, neuropathologic evidence reveals extensive tau burden remains common among people after amyloid removal. If amyloid removal does not also accomplish tau removal, it would not be expected to prevent the cognitive harm from the already-accumulated tau.
A more plausible interpretation of data to date is that the effect of a specific amyloid reduction on tau and downstream variables will be smaller than the effects of that same amyloid increase due to the natural progression of Alzheimer’s disease. Individuals in trials typically already have tau present. (In fact, the TRAILBLAZER trial of donanemab restricted participation to individuals with intermediate levels of tau). While anti-amyloid antibody drugs such as aducanumab appear also to have an effect on CSF p-tau, which would be indicative of slowed tau accumulation in the brain, once tau is seeded, this process may independently continue, resulting in neurodegeneration even if amyloid is reduced.
Ideally, a mathematical model would incorporate our uncertainty about the effect of amyloid on tau and downstream variables. As I’ve argued, this might range from close to no effect of amyloid reduction on tau to an effect equal in magnitude to the amyloid increases on tau obtained from observational studies, which is what I gather was assumed for the Q-ATN model. The lower end of the range—minimal effect of amyloid reduction on tau—does not appear to be represented in the Q-ATN model. If it were, it would likely show that it’s plausible that cognitive benefits would not grow with time.
Ultimately, since a mathematical model is a tool that can be used to show the implications of our assumptions, the validity of the Q-ATN results hinge on the validity of those assumptions. If our input assumptions are inaccurate, our inferences will also be wrong. Therefore, I would argue that since the Q-ATN results depend so heavily on us having an accurate understanding of how to parameterize the underlying biology, they should not be given the same weight as empirical evidence. Ultimately, we will need to collect better data in order to be able to determine the long term-cognitive effects of treatment without having to rely on such potentially unrealistic extrapolations.
References:
Mazer NA, Hofmann C, Lott D, Gieschke R, Klein G, Traber M, Kerchner GA. Linking Amyloid to Cognition in the Pathogenesis and Treatment of Alzheimer’s Disease: Toward the Development of a “Quantitative A/T/N Model” (2239). Neurology Supplement, April 13, 2021 Neurology Supplement .
Nicoll JA, Buckland GR, Harrison CH, Page A, Harris S, Love S, Neal JW, Holmes C, Boche D. Persistent neuropathological effects 14 years following amyloid-β immunization in Alzheimer's disease. Brain. 2019 Jul 1;142(7):2113-2126. PubMed.
Make a Comment
To make a comment you must login or register.