Fluid NfL Shines, Tau PET Dims, in the Hunt for FTD Biomarkers
Quick Links
Biomarkers are the obsession du jour in frontotemporal dementia research. Over and over at the 10th International Conference on Frontotemporal Dementias, held August 31-Septemper 2 in Munich, speakers emphasized the need for new markers to enable research and trials. In a set of diseases bedeviled by both tremendous heterogeneity and small patient populations, biomarkers are must-haves for tracking how disease gets worse and selecting the right participants for trials. In Munich, the consensus was that the field has advanced more on the first goal than the second. Several presentations bolstered the case for the protein neurofilament light (NfL) in blood and cerebrospinal fluid to mark disease stage; NfL also appears to predict future decline. Growing evidence from longitudinal studies suggests that atrophy in key brain areas can flag how close a person is to symptoms. Such biomarkers may help researchers monitor trial participants and perhaps indicate drug effects, although that still has to be tested. “It’s critical to have biomarkers for rational dose finding in Phase 1,” noted Christoph Wiessner of Asceneuron SA, Lausanne, Switzerland.
On the other hand, diagnostic markers that reveal a person’s underlying protein pathology remain elusive. Without them, researchers cannot select the right patients for sporadic FTD trials and instead have to rely on a clinical diagnosis that includes people with similar symptoms but different underlying causes. For example, a person with behavioral variant FTD is about equally likely to harbor tau or TDP-43 pathology, but their clinical symptoms provide no clue which. “We urgently need to develop in vivo biomarkers,” said Ian Mackenzie of the University of British Columbia, Canada. Researchers hoped tau PET might fit the bill, but data at ICFTD made clear that existing tracers cannot discriminate tau and TDP-43 and give but a weak signal from FTD brains. Preliminary data hint that some fluid biomarkers might distinguish between forms of FTD.
Altogether, the research is opening a window into how frontotemporal dementia affects living brains, said Jason Warren of University College London. “The interest in biomarkers represents a paradigm shift for [studying] these diseases. We stand on the verge of a revolution in the treatment of FTD.”
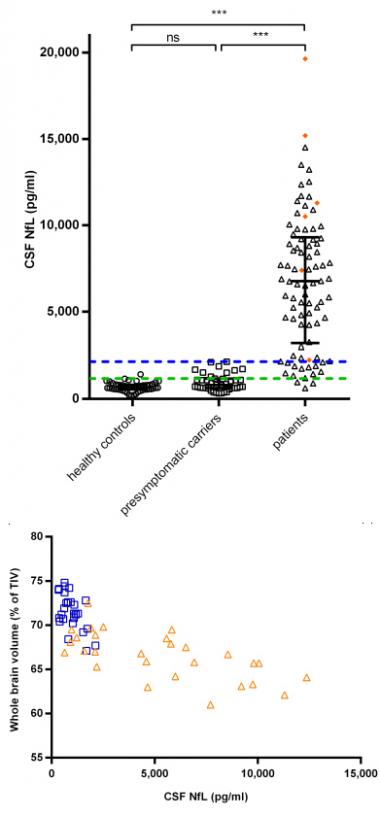
NfL Tracks Degeneration.
CSF levels run high in people with FTD (top) and correlate with brain shrinkage (bottom) in presymptomatic carriers (blue) and FTD patients (orange). [Courtesy of John van Swieten.]
NfL Up and Coming as Progression Marker
Researchers at ICFTD hailed NfL as the most promising FTD biomarker to emerge to date. The protein leaks out of injured axons, and high levels in cerebrospinal fluid and blood appear to reflect the intensity of neurodegeneration in several disorders, including FTD, ALS, and Alzheimer’s disease (see Jun 2015 news; Jun 2016 news). While not specific for disease type, the marker tracks severity quite well, researchers said.
At ICFTD, speakers added new evidence that NfL can also predict progression. John van Swieten of Erasmus Medical Center, Rotterdam, The Netherlands, described findings from 174 Rotterdam participants, most of them in the Genetic Frontotemporal Dementia Initiative 1 (GENFI1). About half of those had symptoms of FTD, with the remainder split between asymptomatic gene carriers and healthy controls. CSF NfL levels in asymptomatic carriers resembled those in controls, while the marker ran eightfold higher in symptomatic participants, van Swieten said. NfL levels correlated with cortical atrophy and a loss of white-matter integrity in the brain (see Meeter et al., 2016). Moreover, in two people who developed symptoms over the course of the study, CSF NfL shot up fourfold over 18 months, suggesting the marker could herald imminent decline. More longitudinal data may help pin down exactly when NfL begins to crest, van Swieten said. In transgenic mice, NfL rises before symptoms appear, he noted. Likewise, data from the Advancing Research and Treatment for Frontotemporal Lobar Degeneration Consortium (ARTFL) also finds differences in NfL levels between asymptomatic carriers and controls, implying the marker precedes symptoms, Brad Boeve of the Mayo Clinic in Rochester, Minnesota, told Alzforum.
While CSF data are informative, clinicians would much prefer a marker they can measure in blood. Here, too, NfL performs well. As other groups had previously, van Swieten reported that serum NfL correlated closely with CSF levels. Julio Rojas of the University of California, San Francisco, added yet more evidence supporting blood measurements. He reported findings from a cohort of 15 people with progressive supranuclear palsy (PSP) and 12 controls. The researchers measured NfL in plasma using a single-molecule array (Simoa), an ultrasensitive technique that employs a digital readout to detect minute quantities (see Apr 2016 conference news). Plasma NfL ran twice as high in cases as controls, with a cutoff of 20 pg/ml providing 80 percent sensitivity and specificity for discriminating the groups, Rojas said (see Rojas et al., 2016).
Rojas and colleagues validated the findings in an independent cohort of 147 PSP patients. Half of this group had very high plasma NfL and half lower, although all were above control levels. Higher NfL predicted worse cognitive, functional, and motor decline one year later, as well as more brain atrophy. Plasma NfL remained stable over that year, and correlated closely with CSF levels. “Tracking blood NfL is feasible, and may support prognosis,” Rojas concluded.
Others at ICFTD reported similar findings for blood measurements. Ione Woollacott of University College London presented data from 74 FTD patients and 28 healthy controls. Serum NfL ran about four times higher in patients than controls. Levels varied widely among individuals, however, and correlated with how quickly their frontal lobe was shrinking (see Rohrer et al., 2016). “NfL is the first good blood biomarker in FTD,” first author Jonathan Rohrer of UCL told Alzforum.
Beyond NfL, researchers are searching for additional fluid markers of progression. MicroRNAs have been found to change in frontotemporal lobar degeneration, and the pattern can vary based on the subtype of disease or the underlying genetics (see Kocerha et al., 2011; Freischmidt et al., 2014). In Munich, Maria Carmela Tartaglia of the University of Toronto reported finding seven microRNAs that were reduced in CSF in 16 people with familial FTLD compared with 19 presymptomatic carriers and seven controls in the GENFI1 cohort. Most carried progranulin mutations. The microRNAs appear to regulate molecular pathways that previously have been implicated in this form of the disease, she told the audience. In a similar vein, van Swieten described a mass spectrometry study of CSF from two Dutch progranulin families. He turned up multiple proteins that distinguished between symptomatic and asymptomatic carriers, including four complement factors and two synaptic proteins. All of these preliminary markers remain to be validated.
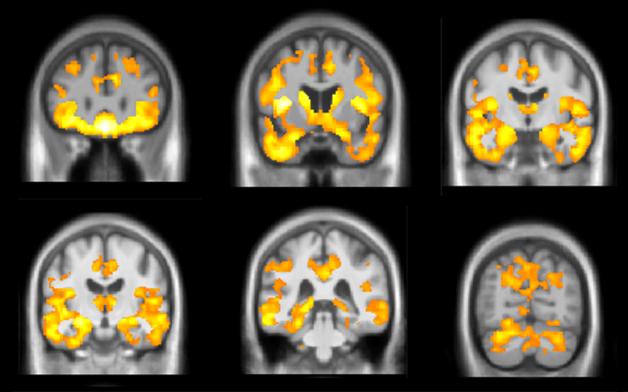
Brain Atrophy Marks Progression. In the GENFI genetic cohort, people who carry a C9ORF72 expansion show accelerated atrophy (yellow) in some brain regions up to 10 years before developing symptoms. [Courtesy of Jonathan Rohrer.]
Structural Imaging May Flag Progression Too
What about brain volumetric imaging? Could it pick out the people most at risk of progressing to FTD? Early indications at ICFTD suggest yes. Martina Bocchetta of UCL presented MRI data from 56 asymptomatic mutation carriers in the GENFI1 cohort. Those who were more than 10 years away from their expected age of onset resembled age-matched controls. However, the brain began to shrink in people within 10 years of their expected onset age. In particular, the researchers saw rates of about 1 percent decline per year in the temporal lobe, cingulate, amygdala, and striatum in this group (see image above). In controls of the same age, these regions remained stable. The findings suggest there might be an atrophy signature that could pick out the people closest to progressing, who might be the best candidates for a clinical trial. “We may be able to use structural imaging [as a biomarker] in trials at that late preclinical period,” Rohrer, who led the study, told Alzforum.
A different progression marker came from the work of Lize Jiskoot, also of Erasmus Medical Center. She analyzed four years of MRI and clinical data from 25 progranulin mutation carriers, nine MAPT mutation carriers, and 29 controls in the Rotterdam Risk Cohort, which is part of GENFI. All were asymptomatic at the start, and thus far three of each genotype have developed FTD symptoms over the course of the study. The researchers saw the cortex shrink faster in MAPT than in GRN carriers. In addition, the four people who developed bvFTD, but not the two who were diagnosed with progressive non-fluent aphasia, lost white matter in the right uncinate fasciculus before symptoms developed, regardless of genotype. This tract connects frontal portions of the salience network, which becomes disrupted in FTD (see Nov 2014 conference news). The findings suggest there may be mutation- and diagnosis-specific atrophy patterns, Jiskoot said.
Howard Rosen of UCSF took a different tack. Instead of presymptomatic carriers, he analyzed MRI images obtained over two years from 44 people with clinical diagnoses of bvFTD, 30 with semantic-variant primary progessive aphasia, and 26 with non-fluent variant primary progressive aphasia, as well as 97 age-matched controls. He asked which brain regions changed the most over time in each disorder and found a different pattern in each. The bvFTD group showed shrinkage in the anterior caudate, insula, and medial frontal and parietal cortex, people with nfPPA lost volume in the perirolandic and periinsular cortices, and those with svPPA in the inferior temporal lobe (see Pankov et al., 2016). An analysis found that these regions detected longitudinal change better than either more commonly used brain regions or clinical measures, Rosen said. If the findings hold up in independent samples, researchers using these regions might need fewer participants in drug trials to see a significant effect, he believes.
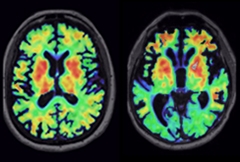
Tau PET Varies by Diagnosis.
PET tracer AV1451 lights up different brain regions in a person with bvFTD caused by a MAPT mutation (right) than in a person with corticobasal degeneration (left), matching expected patterns of tau deposition. [Courtesy of Richard Tsai.]
Fading Hope for Tau PET Leaves Differential Diagnosis Difficult as Ever
While progression markers show promise, the field appears stalled for the moment on its goal of biomarkers that reveal an FTD patient’s underlying pathology. Without that, researchers will remain unable to run effective clinical trials in sporadic bvFTD, said conference co-organizer Manuela Neumann of the German Center for Neurodegenerative Diseases (DZNE). She noted that a clinical diagnosis of bvFTD is wrong half of the time. Even when correct, people with this diagnosis are equally likely to have tau or TDP-43 deposits, and some can have yet other pathologies. FTD researchers had hoped tau PET would distinguish these groups, but numerous presentations at ICFTD emphasized that existing tau tracers work poorly for FTLD. “The tau PET data was the biggest disappointment of the conference,” said Christian Haass of Ludwig-Maximilians University, Munich, expressing a common sentiment.
Existing tracers bind well to the type of tau deposits found in Alzheimer’s disease, which are composed of paired helical filaments made up of 3R and 4R tau strands. Most tau pathology in FTLD, however, consists of either pure 4R or pure 3R tau and, for unknown reasons, these don’t bind the current tracers well. In Munich, Boeve reported that five people with the N279K MAPT mutation, who are known to make 4R tau, weakly bound the tracer AV1451, with intensities falling within the range of binding in healthy control brains. By contrast, two people with the S305N MAPT mutation, who make 3R + 4R tau tangles, took up tracer at levels comparable to AD patients. In an overview talk, Brad Dickerson of Massachusetts General Hospital noted that the tracer THK5351 gives a slightly higher signal in 4R tauopathies than does AV1451, but still far below that seen in AD.
Moreover, AV1451 and THK5351 bind to things other than tau. They recognize still-unknown targets in the midbrain and basal ganglia in particular, confounding the signal from some types of FTLD that feature neurofibrillary pathology in those regions, such as PSP and corticobasal syndrome (CBS). Even more problematic, existing ligands light up brains that contain TDP-43 deposits. Thus, they cannot discriminate these two forms of the disease, Boeve noted. In addition, in autoradiography studies on postmortem tissue samples from FTLD brain, current tracers bind even more poorly than they do in living tissue. This puzzles researchers. Dickerson called the autoradiography data “underwhelming.”
Are the existing tracers useful at all in FTD? Richard Tsai of the UCSF Memory and Aging Center reported that AV1451 uptake matched the expected patterns of tau or TDP-43 pathology in three bvFTD, 10 PSP, nine CBS, and six PPA patients, as well as six FTD mutation carriers and 24 controls (see image above). Corey McMillan of the University of Pennsylvania, Philadelphia, analyzed data from an AV1451 imaging study sponsored by AV1451’s maker, Avid Radiopharmaceuticals. He reported that an AV1451 uptake cutoff of 1.71 SUVR discriminated between 20 PSP patients and 20 controls with a sensitivity and specificity of 80 percent. However, binding was too weak to allow individual diagnosis, McMillan said, and CBS patients did not take up the tracer at all. Jennifer Whitwell of the Rochester Mayo Clinic reported a different pattern of AV1451 binding in logopenic PPA compared with semantic and agrammatic PPA, suggesting PET scans could help differentiate among the different aphasias. In one of the few studies examining THK5351, Sonja Schönecker of Ludwig-Maximilians University reported that this tracer differentiated 10 PSP patients from healthy controls and correlated with clinical severity.
Overall, the data suggest that AV1451 and THK5351 might be useful in clinical trials to show target engagement and determine dosing, but are probably not helpful diagnostically, concluded Adam Boxer of UCSF during a panel discussion. Other companies such as Merck, Genentech, and Roche are developing tau tracers, but did not present at ICFTD (see Feb 2015 conference news; Aug 2016 conference news).
Better Luck with Fluid Markers?
Could biomarkers in CSF or blood become diagnostic? At ICFTD, presentations hinted at some discriminatory power, but so far no marker has broken through as a leading candidate for the non-genetic forms of FTLD. David Irwin of UPenn probed whether CSF p-tau might do the job. He used data from people with FTLD who had undergone a lumbar puncture during life and donated their brains after death. Among 28 people with tau deposits, CSF p-tau ran about 50 percent higher than it did in 45 people with TDP-43 pathology. The difference became even more significant when Irwin screened out 14 cases with comorbid Alzheimer’s pathology. When combined with an Alzheimer’s screen, CSF p-tau might help distinguish these forms of FTLD during life, Irwin suggested.
Rojas presented similar data, reporting that FTLD patients with autopsy-confirmed TDP-43 deposits had about half the CSF p-tau/total tau ratio, and more than twice as much CSF NfL, as did patients with tau pathology. An NfL readout above 6,757 pg/ml and a tau ratio below 0.28 discriminated the two disorders with more than 80 percent sensitivity and specificity in a cohort of 37 patients, Rojas found.
Others focused on new markers. Charlotte Teunissen of VU University Medical Center, Amsterdam, had previously reported that a combination of the inflammatory markers YKL-40, MFGE8, and catalase discriminated 12 patients with FTLD-TDP from eight patients with FTLD-tau (see Teunissen et al., 2016). In Munich, Marta del Campo in Teunissen's group presented the results of replication in two cohorts, one from Emory University and one from Milan University, containing a total of 30 FTLD-TDP patients, 20 FTLD-tau, 29 controls, and 66 with other dementias. The combined biomarkers distinguished the FTLD-TDP cohort from controls with better than 80 percent sensitivity and specificity, suggesting this might become useful to diagnose this form of the disease.—Madolyn Bowman Rogers
References
News Citations
- Blood Marker May Predict ALS Progression
- Blood NfL Looks Good as Progression and Outcome Marker
- WANTED: Biomarkers for Drug Trials in Frontotemporal Dementia
- First Data from GENFI1: Brain’s Insula Region Shrinks A Decade Before FTD
- Human Amyloid Imaging Meeting Was Abuzz With Talk of Tau
- Improving Tau PET: In Search of Sharper Signals
Paper Citations
- Meeter LH, Dopper EG, Jiskoot LC, Sanchez-Valle R, Graff C, Benussi L, Ghidoni R, Pijnenburg YA, Borroni B, Galimberti D, Laforce RJ, Masellis M, Vandenberghe R, Ber IL, Otto M, van Minkelen R, Papma JM, Rombouts SA, Balasa M, Öijerstedt L, Jelic V, Dick KM, Cash DM, Harding SR, Jorge Cardoso M, Ourselin S, Rossor MN, Padovani A, Scarpini E, Fenoglio C, Tartaglia MC, Lamari F, Barro C, Kuhle J, Rohrer JD, Teunissen CE, van Swieten JC. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol. 2016 Aug;3(8):623-36. Epub 2016 Jul 1 PubMed.
- Rojas JC, Karydas A, Bang J, Tsai RM, Blennow K, Liman V, Kramer JH, Rosen H, Miller BL, Zetterberg H, Boxer AL. Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann Clin Transl Neurol. 2016 Mar;3(3):216-25. Epub 2016 Feb 1 PubMed.
- Rohrer JD, Woollacott IO, Dick KM, Brotherhood E, Gordon E, Fellows A, Toombs J, Druyeh R, Cardoso MJ, Ourselin S, Nicholas JM, Norgren N, Mead S, Andreasson U, Blennow K, Schott JM, Fox NC, Warren JD, Zetterberg H. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016 Sep 27;87(13):1329-36. Epub 2016 Aug 31 PubMed.
- Kocerha J, Kouri N, Baker M, Finch N, Dejesus-Hernandez M, Gonzalez J, Chidamparam K, Josephs KA, Boeve BF, Graff-Radford NR, Crook J, Dickson DW, Rademakers R. Altered microRNA expression in frontotemporal lobar degeneration with TDP-43 pathology caused by progranulin mutations. BMC Genomics. 2011;12:527. PubMed.
- Freischmidt A, Müller K, Zondler L, Weydt P, Mayer B, von Arnim CA, Hübers A, Dorst J, Otto M, Holzmann K, Ludolph AC, Danzer KM, Weishaupt JH. Serum microRNAs in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2015 Sep;36(9):2660.e15-20. Epub 2015 Jun 9 PubMed.
- Pankov A, Binney RJ, Staffaroni AM, Kornak J, Attygalle S, Schuff N, Weiner MW, Kramer JH, Dickerson BC, Miller BL, Rosen HJ. Data-driven regions of interest for longitudinal change in frontotemporal lobar degeneration. Neuroimage Clin. 2016;12:332-40. Epub 2015 Aug 18 PubMed.
- Teunissen CE, Elias N, Koel-Simmelink MJ, Durieux-Lu S, Malekzadeh A, Pham TV, Piersma SR, Beccari T, Meeter LH, Dopper EG, van Swieten JC, Jimenez CR, Pijnenburg YA. Novel diagnostic cerebrospinal fluid biomarkers for pathologic subtypes of frontotemporal dementia identified by proteomics. Alzheimers Dement (Amst). 2016;2:86-94. Epub 2016 Jan 19 PubMed.
Other Citations
External Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.