Meet the Switching Mice: They Flip Their Glia APOE4 to APOE2
Quick Links
Is a person born with APOE4 stuck with the lifelong consequences of this risk allele? These days, yes. In the future, perhaps not. APOE4 can be silenced, and the protective APOE2 allele introduced instead, according to Lance Johnson, University of Kentucky, Lexington. Johnson tested this theory by creating APOE4-to-2 “switch” mice. These animals express APOE4 until activation of Cre recombinase shifts expression to APOE2.
- Scientists made mice that switch from expressing ApoE4 to E2 in midlife.
- They limited this swap to microglia or astrocytes.
- After the switch, gene expression reverted to that of mice born with APOE2.
- The astrocyte switch cleared plaques, restored memory.
“This is a very useful model to address the potential benefits of switching E4 to E2 systemically or in a cell-type-specific manner and at different stages of disease,” wrote Ling Li of the University of Minnesota, Minneapolis.
At the Alzheimer’s Association International Conference, held in Amsterdam from July 16-20, Johnson reported that an astrocyte-specific switch cleared plaques, calmed microglia, and improved memory, even when amyloidosis was in full swing. A switch in microglia evoked no such improvements, suggesting that microglial ApoE is unable to stir a response to plaques.
Bart De Strooper, Dementia Research Institute, London, and Amaia Arranz from the Achucarro Basque Center for Neuroscience, Spain, came to a similar conclusion. They reported that astrocyte-derived ApoE rouses other glia against amyloid, whereas microglial ApoE does not. These findings help researchers understand the consequences of someday changing APOE expression in people through gene therapy or gene editing, both of which are being explored.
“After amyloid and tau, I envision APOE could be the next therapeutic target for AD,” said Julia TCW of Boston University, during a lively Q&A. Johnson summed up the potential impact this way: “If we were to gene edit all homozygous APOE4 alleles to APOE2/2, we would prevent 16 percent of AD cases. Changing all heterozygous APOE4 alleles to APOE2 would prevent 63 percent of cases.”
To weaken APOE4, scientists have silenced the gene with antisense oligonucleotides or knocked down the protein with antibodies. Both reduced plaque load in mice, and the latter improved spatial memory (Huynh et al., 2017; Liao et al., 2014; reviewed by Yang et al., 2021). In neurons derived from human induced pluripotent stem cells, CRISPR editing of APOE4/4 to APOE3/3 decreased production of Aβ and phosphorylated tau (Jun 2018 news).
Recent genetic evidence supports the idea that APOE knockdown might work in people, too. One man who has a loss-of-function variant in his APOE4 allele had no amyloid pathology or signs of dementia in his 90s, while a second in his 70s was similarly protected (Aug 2023 news).
Other groups have used gene therapy to bolster APOE2 rather than knock down E4. This decreased levels of soluble oligomeric Aβ and amyloid plaques in mice (Zhao et al., 2016). Likewise, in a first for the field, cerebrospinal fluid phospho-tau and total tau, markers of AD pathology, dropped in three women who are homozygous for APOE4 after a single intrathecal injection of LX1001, an adeno-associated virus carrying the APOE2 gene (Dec 2022 conference news).
Still, there may be downsides to knocking down APOE4 or boosting APOE2. The former might rob the brain of physiological, lipid-toting duties, while adding ApoE2 still leaves ApoE4 running amok. What if both approaches were combined?
With this in mind, Lesley Golden in Johnson’s lab created the APOE switch mice, dubbed 4s2. In these knock-ins, every cell has its mouse APOE gene replaced with a construct that carries genes for human APOE4, and APOE2, with a stop codon in between. The mouse also has an inducible gene for Cre recombinase, which recognizes sites flanking the E4 gene and the stop codon. How does the switch work? The animals make ApoE4 until they are fed tamoxifen. This induces Cre, which then snips out the APOE4, and the stop, freeing up expression of the APOE2 gene (see image below).
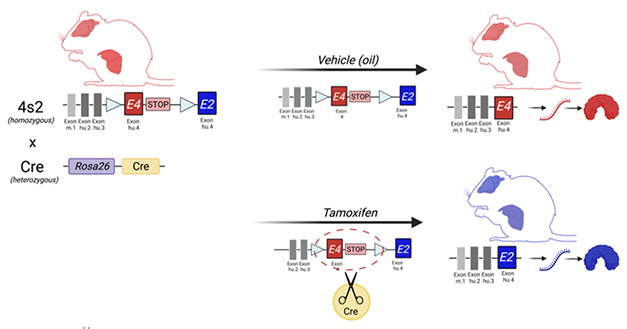
Gene Swap. When APOE4-to-2 (4s2) “switch” mice (left) ingest tamoxifen (right, bottom), Cre recombinase snips out E4, allowing expression of APOE2. 4s2 mice injected with a control (top) kept expressing APOE4. [Courtesy of Lance Johnson, University of Kentucky.]
A month after intraperitoneally injecting 6-month-old 4s2 mice with five daily doses of tamoxifen, Golden detected an almost complete shift in ApoE production. As measured by mass spectrometry, 93 percent and 80 percent of ApoE in the plasma and brain, respectively, was E2.
What happened after the switch? Using single-cell RNA sequencing, the researchers compared gene expression before and after. Switched brain cells resembled cells from APOE2 mice. They expressed many of the same genes unique to E2 cells. For example, 94 percent of the genes expressed by switched astrocytes were APOE2 astrocyte genes. The same was true for 64 percent of microglial genes. “That most of those genes were the same suggests that we can correct most of the differences inherent in APOE4 versus E2 brains with an allele switch in midlife,” Johnson concluded.
Notably, many of these genes support cellular pathways involved in AD, despite these mice having no AD pathology. For example, after the switch, astrocytes and microglia upregulated genes related to lipid metabolism and immune responses, while downregulating genes involved in Aβ precursor protein processing. Some of the downregulated genes had been identified in AD genome-wide association studies, including APOE, microglial-activating cystatin CST3, microglial phagocytosis receptor AXL, and apoptosis-related clusterin.
The switch had a downside, though. Too many triglycerides built up in the blood. A similar disorder, called hyperlipoproteinemia Type 3, occurs in 10 percent of APOE2/2 carriers. It can cause heart attacks or strokes.
To skirt such side effects, Golden used Cre recombinase genes with astrocyte- or microglial-specific promoters to create specific switches. Again, this worked, with 75 percent of astrocytes or microglia expressing ApoE2 after tamoxifen treatment. The scientists have not yet tested if either model developed hyperlipidemia.
Because these astrocyte- and microglia-specific switch mice have no AD pathology, Golden crossed them with 5xFAD mice. The latter begin accumulating plaques and developing gliosis at 2 months of age; spatial memory deficits follow four months later. To see the effect of switching APOE alleles after plaques are established, Golden injected 6-month-old crosses with tamoxifen, then two months later compared their behavior and brains to controls.
The astrocyte-switched mice had half as many amyloid plaques as unswitched mice, but only one-third as many activated astrocytes and microglia (see image below). They did better on memory tests, freezing 40 percent longer when expecting a foot shock. “Astrocyte ApoE influences other cell types involved in AD processes and can drive improvements in pathology and cognition,” Golden concluded.

Potent Astrocyte. Compared to brains of “unswitched” APOE4 5xFAD mice (top left), those whose astrocytes express E2 had fewer amyloid plaques and less gliosis (top middle). Switching APOE in microglia caused a small but non-significant drop in plaques yet increased gliosis (top, right, and inset, bottom). [Courtesy of Lance Johnson, University of Kentucky.]
The microglia switch mice told a more nuanced tale. After tamoxifen, they had no significant change in plaque load, astrogliosis, or memory. Microgliosis worsened. Because microglia produce but a small fraction of the brain's ApoE, switching them barely affects overall ApoE4 production, Johnson suggested. “In the astrocyte-specific switch mice, there is sufficient astrocyte E2 to regulate microglial responses, while in the microglial-specific switches, the astrocyte E4 still outweighs the microglia-derived E2,” he explained.
Similarly, De Strooper and Arranz reported that microglial-derived APOE was unable to rally glia against amyloid, yet APOE from astrocytes did. When Renzo Mancuso was in De Strooper’s lab, he transplanted human microglia into APP NL-G-F mice with or without endogenous APOE. In mice that expressed endogenous ApoE, the human microglia reacted to plaques by shifting from homeostatic to multiple different subtypes. Without murine APOE, the human microglia only mustered the cytokine-producing subtype (Oct 2022 news). “We speculate that the APOE secreted from microglia is a weak modulator of their response,” said De Strooper.
In Amsterdam, Arranz reported that she has now created similar mouse chimeras, but with astrocytes. She transplanted human astrocytes expressing APOE3 or E4 into one brain hemisphere of newborn APP/PS1 mice. Six months later, the side of the brain hosting human E3 cells had fewer amyloid plaques, with fewer microglia surrounding them, than did the contralateral side. In contrast, hemispheres injected with E4 astrocytes had more plaques than the contralateral side, and the plaques swarmed with microglia. “These data support the idea that human astrocytes modify microglial responses to AD pathology and might have a more impactful role than previously thought,” Arranz said.
All told, the three presentations support the idea that astrocyte-APOE potently regulates glial response to AD pathology, suggesting that future therapies should focus on modifying APOE alleles in those cells.
If anyone can pull off such an APOE4-E2 switch in people, then when during the AD continuum would be the time to do it? Before or after plaques have begun to accumulate? Golden is testing if giving tamoxifen early, i.e., at 2 months, will prevent AD pathology in the 5xFAD/4s2 mice. Johnson plans to cross the 4s2 mice with tauopathy models.
Other labs may soon join in. The wild-type 4s2 mouse will be available through Jackson Laboratory (JAX), Bar Harbor, Maine, later this year. Mike Sasner and colleagues at JAX are creating three other switch mice—APOE3-to-2, APOE4-to-3, and APOE3-to-4, so scientists can assess different allele-swapping scenarios. These mice will be available in mid-2024. Scientists in Amsterdam said they are looking forward to using these models.
Could an APOE switch be therapeutic? Amila Zuko and colleagues at UniQure Biopharma in Amsterdam are testing a dual, APOE4 knock-down/gene therapy approach in mice. Into the striata of wild-type animals they injected a virus containing both a microRNA to suppress APOE4 expression and a gene to overexpress a protective form of APOE, which they did not disclose. At AAIC, the scientists reported that a month after injection, the microRNA and the protective ApoE were produced at high levels in the cortex. In mice expressing human APOE4, injecting just the microRNA knocked down the gene by 60 percent. Zuko's next step is to test the dual therapy in APOE4 mice. This approach is not cell-specific. It is targeted to the brain by direct injection, à la LX1001 in the APOE2 gene therapy trial.—Chelsea Weidman Burke
References
Mutations Citations
News Citations
- In Human Neurons, ApoE4 Promotes Aβ Production and Tau Phosphorylation
- Goodbye, APOE4. Hello, Healthy Brain?
- In Small Trial, Gene Therapy Spurs ApoE2 Production
- Human Microglia Mount Multipronged Response to AD Pathology
Therapeutics Citations
Research Models Citations
Paper Citations
- Huynh TV, Liao F, Francis CM, Robinson GO, Serrano JR, Jiang H, Roh J, Finn MB, Sullivan PM, Esparza TJ, Stewart FR, Mahan TE, Ulrich JD, Cole T, Holtzman DM. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of β-amyloidosis. Neuron. 2017 Dec 6;96(5):1013-1023.e4. PubMed.
- Liao F, Hori Y, Hudry E, Bauer AQ, Jiang H, Mahan TE, Lefton KB, Zhang TJ, Dearborn JT, Kim J, Culver JP, Betensky R, Wozniak DF, Hyman BT, Holtzman DM. Anti-ApoE antibody given after plaque onset decreases Aβ accumulation and improves brain function in a mouse model of Aβ amyloidosis. J Neurosci. 2014 May 21;34(21):7281-92. PubMed.
- Yang A, Kantor B, Chiba-Falek O. APOE: The New Frontier in the Development of a Therapeutic Target towards Precision Medicine in Late-Onset Alzheimer's. Int J Mol Sci. 2021 Jan 27;22(3) PubMed.
- Zhao L, Gottesdiener AJ, Parmar M, Li M, Kaminsky SM, Chiuchiolo MJ, Sondhi D, Sullivan PM, Holtzman DM, Crystal RG, Paul SM. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer's disease mouse models. Neurobiol Aging. 2016 Aug;44:159-72. Epub 2016 Apr 30 PubMed.
Further Reading
Follow-On Reading
Papers
- Golden LR, Siano DS, Stephens IO, MacLean SM, Saito K, Nolt GL, Funnell JL, Pallerla AV, Lee S, Smith C, Chen J, Zhu H, Voy C, Whitus CM, Hernandez G, Farmer BC, Pandya K, Cowley DO, Macauley SL, Gordon SM, Morganti JM, Johnson LA. APOE4 to APOE2 allelic switching in mice improves Alzheimer's disease-related metabolic signatures, neuropathology and cognition. Nat Neurosci. 2025 Dec;28(12):2461-2475. Epub 2025 Nov 11 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Arkansas for Medical Sciences
This new model is intriguing for its potential to swing the pathogenic activities of ApoE in such dramatic extremes. This should create a strong contrast between the downstream consequences, such as profiles of gene expression. But one could achieve that through a simple comparison of ApoE2-expressing tissue to ApoE4-expressing tissue. The beauty of an induced switch like this is really the ability to mimic a naturally occurring change that occurs over ontological development or disease development. There is no equivalent to an E4-to-E2 switch that occurs in nature. For this reason, I do not expect this model to provide major benefits for translational projects or clinical trials. I am unaware of any trials that aim to implement such a dramatic gene therapy feat.
One aspect of the model that provides a useful leverage point is the ability to manipulate the ApoE variant in a specific cell type. As a secreted protein, ApoE might be expected to operate independently of the cell type expressing it, but our group is testing a hypothesis that seems to hinge on cell-autonomous actions of ApoE.
The amelioration of pathology observed when the E4-to-E2 conversion occurred in astrocytes, but not microglia, is consistent with results obtained with Oleg Butovsky’s models, wherein beneficial effects in an AD model followed removal of ApoE4 expression from astrocytes instead of microglia. The interpretation is that ApoE4 may promote a form of activation in microglia that facilitates Aβ clearance.
University of Minnesota, Twin Cities
It was great to hear the talks from Lance Johnson and graduate student Lesley Golden on the APOE4-to-APOE2 “switch mouse” (APOE4s2) model at AAIC in Amsterdam. Although isogenic APOE iPSCs have been established in vitro using the CRISPR gene editing technology, this is the first inducible mouse model in which E4 can be switched to E2 systemically or in a cell-type-specific manner at different ages/stages of disease development.
Golden et al. showed that systemic switch of E4 to E2 led to a phenotype similar to that of the E2-TR mice, including hyperlipidemia/hypertriglyceridemia. In the brain, as expected, APOE originated primarily from astrocytes, with some from microglia. Excitingly, the astrocyte-specific switch from E4 to E2 reduced Aβ load and gliosis, and improved associative memory in 5xFAD mice that had already developed robust amyloid pathology. In contrast, a microglia-specific switch from E4 to E2 did not produce the same effects.
These findings indicate that astrocytic E2 expression is critical for the beneficial outcomes, and microglial E2 expression is insufficient to counteract the effects of E4. However, APOE expression in microglia is much lower than in astrocytes. Whether increasing the microglial E2 expression to a similar level as in astrocytes could lead to comparable effects is not known.
Notably, E4 carriers have lower levels of APOE compared to E2 carriers, a difference that was recapitulated in the APOE4s2 mice, most likely due to poor lipidation state and faster turnover rate of E4 (Hanson et al., 2013; Kanekiyo et al., 2014). Therefore, it is unclear whether the level/lipidation state of APOE or APOE2 per se is important for the modulation of AD progression. We and others have shown that enhancing lipidation of APOE restores E4 function and ameliorates AD-related pathology (Boehm-Cagan et al., 2016; Chernick et al., 2018; Apr 2019 conference news).
Therapeutically, it is worth noting that, in addition to the undesirable phenotype of hyperlipidemia, E2 has also been associated with increased tau pathology in primary tauopathy (Zhao et al., 2018). What effect the E4-to-E2 switch may have on AD-type tau pathology awaits further investigation.
Despite these concerns and questions, the protective effect of E2 against AD risk is strongly supported by genetic studies. The APOE4s2 mouse provides a unique in vivo model to assess the therapeutic potential of E2 replacing E4 in a temporal and cell-specific controlled manner.
Lastly and importantly, I would like to applaud Lance for sharing with the audience at AAIC, for donating this mouse line to JAX Repository and making it available to the global biomedical research community. This certainly will help accelerate the understanding of pathophysiological outcomes of the E4 to E2 switch.
References:
Boehm-Cagan A, Bar R, Liraz O, Bielicki JK, Johansson JO, Michaelson DM. ABCA1 Agonist Reverses the ApoE4-Driven Cognitive and Brain Pathologies. J Alzheimers Dis. 2016 Oct 4;54(3):1219-1233. PubMed.
Chernick D, Ortiz-Valle S, Jeong A, Swaminathan SK, Kandimalla KK, Rebeck GW, Li L. High-density lipoprotein mimetic peptide 4F mitigates amyloid-β-induced inhibition of apolipoprotein E secretion and lipidation in primary astrocytes and microglia. J Neurochem. 2018 Dec;147(5):647-662. Epub 2018 Nov 26 PubMed.
Hanson AJ, Bayer-Carter JL, Green PS, Montine TJ, Wilkinson CW, Baker LD, Watson GS, Bonner LM, Callaghan M, Leverenz JB, Tsai E, Postupna N, Zhang J, Lampe J, Craft S. Effect of apolipoprotein e genotype and diet on apolipoprotein e lipidation and amyloid peptides: randomized clinical trial. JAMA Neurol. 2013 Aug 1;70(8):972-80. PubMed.
Kanekiyo T, Xu H, Bu G. ApoE and Aβ in Alzheimer's disease: accidental encounters or partners?. Neuron. 2014 Feb 19;81(4):740-54. PubMed.
Zhao N, Liu CC, Van Ingelgom AJ, Linares C, Kurti A, Knight JA, Heckman MG, Diehl NN, Shinohara M, Martens YA, Attrebi ON, Petrucelli L, Fryer JD, Wszolek ZK, Graff-Radford NR, Caselli RJ, Sanchez-Contreras MY, Rademakers R, Murray ME, Koga S, Dickson DW, Ross OA, Bu G. APOE ε2 is associated with increased tau pathology in primary tauopathy. Nat Commun. 2018 Oct 22;9(1):4388. PubMed.
Make a Comment
To make a comment you must login or register.