From Phagocytosis to Exophagy: Microglia's Digestive Tract Dissected
Quick Links
The scientific diet at this year’s International Conference on Alzheimer’s and Parkinson’s diseases, March 28 to April 1 in Gothenburg, was especially rich in news about the ins and outs of the microglial “gastrointestinal tract,” aka their endolysosomal system. From binging to indigestion to purging, research on microglial peristalsis, essentially, moved scientists' understanding on cellular and molecular mechanisms in neurodegenerative disease.
- Using “lysosomal synapses,” microglia digest extracellular plaques—and inadvertently spread them.
- In familial British amyloidosis, microglia themselves were fingered as the source of the disease's amyloid.
- Within the lysosomes of microglia, Aβ and tau aggregates age and grow.
- From a “village” of microglia, TMEM106b links AD risk to poor phagocytosis.
Results converged largely on the lysosome. Sensing Aβ aggregates, microglia reportedly send these digestive sacs to the cell surface, where they dump their enzyme juices onto plaques. But Aβ aggregates gave some microglia a sour stomach. They festered in their lysosomes, growing larger over time, ready to contribute to the spread of amyloidogenic seeds when spewed out later. TMEM106b, a lysosomal protein with genetic ties to several neurodegenerative diseases, connected the dots between AD risk and floundering microglial phagocytosis (see Part 14 of this series). In all, the findings appear to put malfunctions in microglial lysosomes at the heart of the early, cellular phase of neurodegenerative disease.
Microglia are intertwined with every stage of amyloid plaque development, from construction to containment to clearance. It is clear that the cells engulf Aβ aggregates via phagocytosis, but how do they manage to consume plaques that are several times their size? Unlike a snake that unhinges its jaw bones when eating animals larger than itself, microglia might well digest plaques without eating them at all. At AD/PD, Santiago Sole-Domenech, a research associate in Frederick Maxfield’s lab at Weill Cornell Medical College in New York, showed how that could work.
Graduate student Rudy Jacquet and Sole-Domenech took a cue from the way macrophages digest large LDL particles, or osteoclasts reabsorb bone. Maxfield’s group previously showed that, using a process called digestive exophagy, these other myeloid cells direct their lysosomes to the cell surface, where these vesicular troops fuse with the plasma membrane, hurling the brew of enzymes within them onto their extracellular prey (Singh et al., 2016). Could microglia use the same technique to degrade Aβ aggregates?
Jacquet found that when exposed to fluorescently labeled aggregates of Aβ, primary microglia quickly rearranged their plasma membranes to form an invagination surrounding each aggregate.
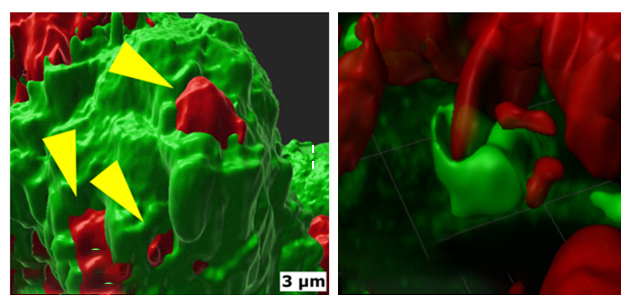
Digestive Hug. Exposure to Aβ aggregates (red) triggered microglia to rearrange their cytoskeleton (green) and wrap the aggregates in pockets of plasma membrane. [Courtesy of Santiago Sole-Domenech, Weill Cornell Medical College.]
Using pH-sensitive dyes, the scientists found that microglia released acidic, lysosomal contents into these aggregate-clutching pockets, a phenomenon Maxfield had christened as “lysosomal synapses.” They only formed in regions of the membrane in direct contact with Aβ aggregates. The same phenomenon appeared to occur in the brain. Using electron microscopy of brain slices from 5xFAD mice, the researchers spotted microglia forming membrane pockets right where they touched plaques. These extracellular compartments brimmed with acid phosphatase, an abundant lysosomal enzyme.

Lysosomal Synapses. A microglial cell (gray) sits underneath an Aβ aggregate (red). At the point of contact, it released acidic contents of its lysosomes, as detected by pH-sensitive dye (green, arrowhead). [Courtesy of Santiago Sole-Domenech, Weill Cornell Medical College.]
While this external digestion may initially shrink plaques, it could potentially backfire, Sole-Domenech reported. He found that when microglia were first allowed to gorge themselves on small Aβ fibrils before encountering large aggregates, the cells filled up their lysosomes with fibrils, then later spewed them out onto large Aβ aggregates via lysosomal synapses.
Akin to the way the process is regulated in LDL-digesting macrophages, Sole-Domenech and Maxfield found that signaling via MyD88, downstream of toll-like receptor 4 and CD14 receptors, instigated digestive exophagy in microglia.
How might signaling through TREM2, a microglial receptor that promotes phagocytosis of Aβ aggregates, influence this external digestion? Strikingly, Jacquet and Sole-Domenech found that microglia deficient in TREM2 doubled down on digestive exophagy, unleashing even more lysosomal contents onto Aβ plaques. He hypothesized that TREM2-deficient microglia might ramp up these external digestive mechanisms to compensate for their deficits in phagocytosis. In particular, TREM2-deficient microglia poorly polymerize actin, and phagocytosis requires more actin polymerization than does digestive exophagy, Sole-Domenech told Alzforum. “It is therefore ‘easier’ for the cells to exocytose lysosomal contents during digestive exophagy,” he added. In support of this idea, he said wild-type and TREM2-deficient microglia form lysosomal synapses with equal measure when they are treated with GM-CSF, which boosts actin polymerization.
In all, Sole-Domenech cast digestive exophagy as a double-edged sword: It allows microglia to cut Aβ plaques down to size without internalizing them, but if microglia also fill themselves with smaller Aβ aggregates via phagocytosis, they run the risk of regurgitating—and potentially spreading—undigested toxic amyloidogenic peptides.
This exact flavor of microglial indigestion was the topic of Anja Schneider’s talk at AD/PD. Schneider, of the German Center for Neurodegenerative Diseases in Bonn, used an isotope-labeling technique to track the evolution of aggregates within the mouse brain. When 5xFAD mice reached 6 months of age, she swapped out their regular chow with food containing 13C-labeled lysine. This heavy isotope can be distinguished from the lighter 12C variety using nanoSIMS, a form of mass spectrometry that can track isotopes with nanoscale resolution. Schneider was then able to distinguish between relatively younger and older proteins within the mouse brain.
Schneider found that roughly 30 percent of Aβ aggregates resided inside of cells. Microglia stomached most of that intracellular pool within their lysosomes. Isotope scanning of the lysosomal aggregates revealed that Aβ peptides in the core were substantially older than those in the outer shell, suggesting that fresh Aβ peptides were continually joining up with older aggregates that were sitting within the lysosome.
The results took Schneider by surprise. “We had assumed that Aβ aggregates are being degraded in the lysosome, not that they grow there.”

Old at Heart. In Aβ aggregates within microglial lysosomes, the oldest peptides (red) accumulate in the core, with progressively younger peptides forming the shell. [Courtesy of Anja Schneider, DZNE, Bonn]
Schneider came to similar conclusions about tau. This protein accumulated within microglial lysosomes of P301L mice and, once again, younger tau proteins surrounded a comparatively ancient aggregate core. Schneider proposed that instead of clearing amyloid or tau aggregates, microglial phagocytosis might sometimes lead to the formation of amyloidogenic seeds. If the contents of these lysosomes were to be secreted, say, by a mechanism like digestive exophagy, this could lead to propagation rather than containment. Schneider and Sole-Domenech agreed that their findings complement each other, and are consistent with the hypothesis that buildup of aggregates within lysosomes could ultimately contribute to amyloid dissemination. In light of these findings, Schneider believes the field should approach therapies aiming to cautiously increase microglial phagocytosis.
Marc Diamond of UT Southwestern Medical Center questioned the conclusion that aggregates were actually growing within the lysosomes. He posed an alternative explanation, that aggregates were fully formed—with old proteins in the core and young ones in the shell—prior to their phagocytosis. Schneider acknowledged that possibility, adding that one would need to label proteins at different time points to determine exactly when and where the aggregates formed. Whether the aggregates grew in the lysosomes or not, the findings do suggest that microglia fail to degrade them.
What prevents microglia from fully digesting Aβ aggregates within their lysosomes? Schneider and Sole-Domenech noted that the acidic pH of late endosomes and lysosomes provide the optimal environment for Aβ aggregation. “We think that low pH and high local intralysosomal concentrations may favor aggregate formation,” Schneider said.
The AD/PD meeting was not the first time microglia have been painted as disseminators of amyloid. Previous studies have implicated the cells in the propagation of tau pathology, reporting that microglia readily consume tau aggregates but fail to destroy them, leading to their eventual release (Jul 2018 conference news; Wang et al., 2022; Hopp et al., 2018). In a variation on this theme, studies from Seiko and Tsuneya Ikezu’s lab, then at Boston University School of Medicine and now at Mayo Clinic Jacksonville, Florida, reported that microglia release tau aggregates encased in extracellular vesicles (Clayton et al., 2021).
At AD/PD, Seiko Ikezu expanded on these findings. She described what happened when microglia were deprived of TSG101, a member of the ESCRT-I complex that forms extracellular vesicles. In conditional knockout mice, removing TSG101 from microglia stymied the propagation of tau tangles from the entorhinal cortex into the hippocampus, a propagation route the researchers had previously described (Sep 2021 news). In a P301S mouse model of tauopathy, nixing TSG101 from microglia roughly halved the burden of tau tangles in the hippocampus, and rescued memory deficits.
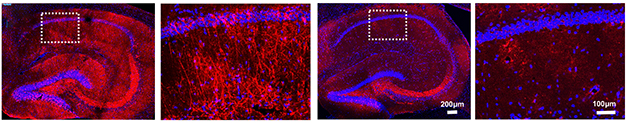
No Transport for Tau. Misfolded tau (red) accumulated in the hippocampi of PS19 mice (left panels). Deletion of TSG101 in microglia cut the tau burden in half (right panels). [Courtesy of Seiko Ikezu, Mayo Clinic.]
Curiously, without TSG101, microglia were less likely to shift into a disease-associated transcriptional state. They expressed less of the complement receptor C3aR1, and munched on fewer synapses than did their TSG101-replete counterparts. In culture, primary microglia sans TSG101 had little appetite for synaptosomes, while microglia from wild-type mice readily consumed them.
The Ikezus’ data indicate that the microglial production of extracellular vesicles exacerbates tau pathology. Exactly how cutting off EV secretion might also counteract microglial activation needs further investigation, Ikezu said. Much as constipation reduces appetite, blocking the release of EVs apparently blocks microglial phagocytosis. Ikezu believes that phagocytosis of apoptotic neurons, damaged synapses, or aggregated proteins is an essential part of the microglial transition into the DAM state, so this could explain how TSG101 deletion ultimately douses microglial activation, she told Alzforum. It is also possible that the EVs contain inflammatory ligands, such as nucleic acids or mitochondrial proteins, that push microglia into the DAM state, she added.
In a startling example of microglia gone terribly wrong, Charles Arber, working in the lab of Selina Wray at University College London reported that the cells are the sole purveyors of the amyloidogenic peptide that causes familial British dementia. This rare genetic disorder is marked by amyloid plaques and tau tangles, but its plaques contain no Aβ. They are made of a peptide cleaved from the C-terminus of the transmembrane protein ITM2B. Mutations that cause FBD disrupt a stop codon in the ITM2B gene, resulting in production of the aggregation-prone, 34-amino acid “A-Bri” peptide.
Together with Sarah Wiethoff, Arber acquired fibroblasts from two people with FBD and two controls, and differentiated them into iPSCs. Try as she might, Emma Augustin from Arber’s lab found no trace of ITM2B, the A-Bri peptide, or any phenotype whatsoever, in iPSC-derived neurons from the two people with FBD. Taking a hint from other gene-expression datasets that suggested ITM2B was expressed in microglia, the scientists differentiated the iPSCs into microglia. Lo and behold, microglia from both control and patient samples expressed ITM2B, and A-Bri was found only in FBD. Microglia were also spotted manufacturing A-Bri in brain samples from a person with FBD, as well as from a person with familial Danish dementia, a related disorder caused by mutations in the same gene.
A dive into single-cell transcriptomic datasets revealed that ITM2B is a disease-associated microglia (DAM) signature gene, rising in step with TREM2, Tyrobp, and other DAM genes. What does this mean? Arber proposed that FBD could be sparked when an inflammatory event provokes microglia to shift into the DAM-like state. In support of this idea, Arber said that many people develop FBD symptoms after experiencing such events, such as a flu infection or a stroke. In other words, in people carrying a causative ITM2B mutation, an otherwise beneficial response to, say, a viral infection or a brain trauma, causes microglia, of all cells, to produce an amyloidogenic protein that leads to dementia.
David Holtzman of Washington University in St. Louis called the production of A-Bri by microglia “an amazingly cool finding. I don’t know of any other extracellular amyloid that is mostly produced by microglia.” Regarding the idea that the disorder could be sparked by neuroinflammation, Holtzman wondered if familial British dementia, like AD, starts with a long preclinical phase where plaques steadily grow. If so, how would one reconcile that long asymptomatic period with the idea that symptoms of disease surface soon after an inflammatory event? Arber said those remain open questions, given the dearth of preclinical samples or biomarkers for this disease. “Still, it is exciting to think that inflammation is part of the onset and progression,” Arber said.
This sampling of microglial behavior on display at AD/PD offers but a glimpse of the myriad ways the cells react to different stimuli. How do these reactions—and the transcriptional states that control them—influence neurodegenerative disease? In the next part of this series, read how scientists grapple with this question by marrying cutting edge omics and human microglial cell culture techniques.—Jessica Shugart
References
News Citations
- With Microglia, It Takes a Village to Connect the Dots
- Synaptic Tau Clangs the Dinner Bell for Hungry Microglia
- Wolframin-1 Cells: Tau’s Launch Pad from Entorhinal Cortex to Hippocampus?
Paper Citations
- Singh RK, Barbosa-Lorenzi VC, Lund FW, Grosheva I, Maxfield FR, Haka AS. Degradation of aggregated LDL occurs in complex extracellular sub-compartments of the lysosomal synapse. J Cell Sci. 2016 Mar 1;129(5):1072-82. Epub 2016 Jan 22 PubMed.
- Wang C, Fan L, Khawaja RR, Liu B, Zhan L, Kodama L, Chin M, Li Y, Le D, Zhou Y, Condello C, Grinberg LT, Seeley WW, Miller BL, Mok SA, Gestwicki JE, Cuervo AM, Luo W, Gan L. Microglial NF-κB drives tau spreading and toxicity in a mouse model of tauopathy. Nat Commun. 2022 Apr 12;13(1):1969. PubMed.
- Hopp SC, Lin Y, Oakley D, Roe AD, DeVos SL, Hanlon D, Hyman BT. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer's disease. J Neuroinflammation. 2018 Sep 18;15(1):269. PubMed.
- Clayton K, Delpech JC, Herron S, Iwahara N, Ericsson M, Saito T, Saido TC, Ikezu S, Ikezu T. Plaque associated microglia hyper-secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Mol Neurodegener. 2021 Mar 22;16(1):18. PubMed. Correction.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.