Salzburg: Protein Trio Offers Handle on Elusive ERAD
Quick Links
No one disputes that Alzheimer’s, as well as other neurodegenerative illnesses, are in part diseases of misfolded protein piling up in neurons. Yet it has been difficult to study exactly where things go awry because some of the parts of the molecular machinery with which neurons dispose of misshapen proteins have been mysterious. At the 8th International Conference AD/PD, held last month in Salzburg, Austria, Mervyn Monteiro of the University of Maryland Biotechnology Institute in Baltimore offered up a new protein complex for the field to study. Monteiro suggested that the new protein erasin forms a platform on which the proteins p97 (aka VCP) and ubiquilin settle down as they yank potential troublemakers out of the lumen of the ER and render them to the proteasome for degradation.
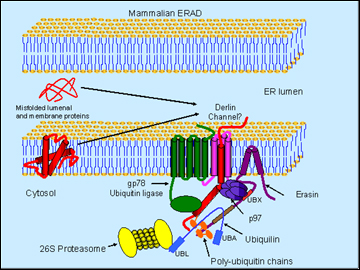
Schematic model of erasin-p97-ubiquilin complex in ERAD (Image credit: Mervyn J. Monteiro)
This proposed garbage crew helps shed new light on a regulated process called ERAD, short for endoplasmic reticulum-associated degradation. ERAD has its own literature going back a decade, but researchers in neurodegenerative diseases have not been able to get a good fix on its molecular players. Monteiro’s new science is of the basic kind, though disease connections for the involved proteins abound. P97 is none other than the valosin-containing protein known to cause rare familial of frontotemporal dementia with ubiquitin and TDP-43-positive inclusions (see ARF Salzburg story), as well as inclusion body myopathy and Paget's disease of bone. Ubiquilin is a presenilin-binding protein found in postmortem tissue of AD brain and in huntingtin aggregates. In research models of triplet-repeat diseases, ubiquilin protects against polyQ-expanded huntingtin and the formation of polyQ inclusions. It is one of many genes for which some genetic association data exist, though as with many candidate genes, other studies to date have not reproduced initial findings (see Alzgene). More detailed study of these proteins may shed light on the common aspects that many protein misfolding diseases share.
Monteiro presented data to suggest that manipulating poly-Q huntingtin in the worm C. elegans supported the idea that it facilitates degradation of this misfolding protein. Ubiquilin overexpression decreased both protein aggregation and a movement phenotype, whereas RNAi knockdown exacerbated these readouts (Wang et al., 2006). Ubiquilin sped up the selective degradation of the expanded huntingtin. But how might it do that? To explain, Monteiro introduced erasin (Liang et al., 2006), an ERAD protein that interacts with ubiquilin. Thanks to a short hydrophobic region, erasin is firmly anchored in the ER while dangling most of its sequence into the cytoplasm, where it exposes two important binding sites. One of those, a UBX domain close to the membrane, binds the p97/VCP; the other one is a coiled-coil motif farther down the protein, which binds ubiquilin.
Here is Monteiro’s working model of this complex in a nutshell: erasin forms a platform that recruits p97/VCP and ubiquilin. The former is a molecular chaperone that extracts misfolded proteins from the ER, possibly through channels called derlins. The trash-to-be gets ubiquitinated by a ubiquitin ligase (such as gp78) as it enters the cytosol. Because the UBA domain of ubiquilin can bind poly-U chains and ubiquitin is positioned conveniently downstream of p97 on the erasin protein, the tagged protein gets handed on to ubiquilin. Then the N-terminal UBL domain of ubiquilin binds the proteasome and delivers the trash into its teeth.
As supporting evidence, Monteiro showed data suggesting that erasin levels shoot up with ER stress and that proteasome inhibition increases ubiquilin-erasin binding. In collaboration with Ralph Nixon at the Nathan Kline Institute in Orangeburg, New York, the scientists showed that erasin antibody staining is increased in AD postmortem tissue. Monteiro’s take-home message is that the complex of erasin, ubiquilin, and p97/VCP helps clear misfolded proteins from the ER.—Gabrielle Strobel.
References
News Citations
Paper Citations
- Wang H, Lim PJ, Yin C, Rieckher M, Vogel BE, Monteiro MJ. Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington's disease by ubiquilin. Hum Mol Genet. 2006 Mar 15;15(6):1025-41. PubMed.
- Liang J, Yin C, Doong H, Fang S, Peterhoff C, Nixon RA, Monteiro MJ. Characterization of erasin (UBXD2): a new ER protein that promotes ER-associated protein degradation. J Cell Sci. 2006 Oct 1;119(Pt 19):4011-24. PubMed.
External Citations
Further Reading
Papers
- Weihl CC, Miller SE, Hanson PI, Pestronk A. Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum Mol Genet. 2007 Apr 15;16(8):919-28. PubMed.
- Ballar P, Shen Y, Yang H, Fang S. The role of a novel p97/valosin-containing protein-interacting motif of gp78 in endoplasmic reticulum-associated degradation. J Biol Chem. 2006 Nov 17;281(46):35359-68. PubMed.
- Thomas AV, Herl L, Spoelgen R, Hiltunen M, Jones PB, Tanzi RE, Hyman BT, Berezovska O. Interaction between presenilin 1 and ubiquilin 1 as detected by fluorescence lifetime imaging microscopy and a high-throughput fluorescent plate reader. J Biol Chem. 2006 Sep 8;281(36):26400-7. PubMed.
- Ford DL, Monteiro MJ. Dimerization of ubiquilin is dependent upon the central region of the protein: evidence that the monomer, but not the dimer, is involved in binding presenilins. Biochem J. 2006 Nov 1;399(3):397-404. PubMed.
- Yang H, Zhong X, Ballar P, Luo S, Shen Y, Rubinsztein DC, Monteiro MJ, Fang S. Ubiquitin ligase Hrd1 enhances the degradation and suppresses the toxicity of polyglutamine-expanded huntingtin. Exp Cell Res. 2007 Feb 1;313(3):538-50. PubMed.
- Hiltunen M, Lu A, Thomas AV, Romano DM, Kim M, Jones PB, Xie Z, Kounnas MZ, Wagner SL, Berezovska O, Hyman BT, Tesco G, Bertram L, Tanzi RE. Ubiquilin 1 modulates amyloid precursor protein trafficking and Abeta secretion. J Biol Chem. 2006 Oct 27;281(43):32240-53. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.