sAPP Binds GABA Receptor, and More News on APP
Quick Links
Researchers convened last month in Heidelberg, Germany, could have been forgiven for thinking they had been teleported back to AD research of the 1990s. Classic, good old biochemistry captured the limelight at Mechanisms in Neurodegeneration, an EMBO/EMBL symposium. Researchers shared new data on the properties and processing of amyloid precursor protein and its fragments. They evoked new insights into γ-secretase function to explain the different potencies of various presenilin mutations and to envision γ-secretase inhibitors of the future that might spare everything bar the amyloidogenic C-terminal fragments of APP. A new candidate for the long-elusive function of APP aired when Bart De Strooper, who co-organized the meeting with Karin Dumstrei, Todd Golde, and Christian Haass, claimed that APP modulates GABA neurotransmitter receptors. Evidence that an astrocytic protease might dispatch up to half the Aβ normally produced in the brain generated a buzz.
Do Astrocytes Degrade Aβ?
Researchers led by Taisuke Tomita, University of Tokyo, homed in on the latter topic when studying Aβ degradation in cell culture. In Heidelberg, Tomita described how adding Aβ-laden conditioned medium from 7PA2 cells to neuron-astrocyte co-cultures led to reduced Aβ levels after a dozen days in vitro. During the same period, astrocytes grew in number, suggesting they may have a role in clearing the peptide. Looking at individual cell types, Tomita found that neither MG6 nor BV2 microglial lines were able to degrade Aβ, but the CCF-STTG1 astrocytoma cell line did. Tomita blocked this degradation with the inhibitor TPCK, hinting a chymotrypsin-like protease was responsible. But which one? Backed by a slew of in vitro and in vivo data that researchers at the meeting called convincing and complete, Tomita made the case that it was kallikrein 7, aka KLK7.
Previously, Patrick Daugherty and colleagues at University of California Santa Barbara had reported that KLK7, a serine protease with a unique chymotrypsin-like activity, cleaved the amyloidogenic core of Aβ (see Shropshire et al., 2013). Those experiments were based on phage display, and heretofore, no one had linked KLK7 to AD pathology, said Tomita.
Using MAB2624, a monoclonal antibody to the protease, Tomita found that astrocytes, but not microglia, expressed KLK7. Using postmortem brain samples from Japanese people aged 60-90, he found there was less of the protein in AD than in control brains. Turning to mice, Tomita asked if the protease affected amyloidosis. He crossed KLK7 knockouts with APP knock-ins created by Takaomi Saido at RIKEN in Wako, Japan (Saito et al., 2014). The crosses had more soluble Aβ in the brain than controls, and 3.5 times as many Aβ plaques. Phosphorylated tau, as determined by AT180, ticked up 20 percent. Interestingly, the KLK7-negative mice had more β-secretase around plaques, and astrocytes were more active than in controls, as well.
Do astrocytes upregulate KLK7 in response to amyloidosis? Tomita found that Aβ boosted expression of KLK7 in astrocyte cultures, suggesting the protease responds homeostatically to local Aβ levels. He looked for other signals that might do the same. Lipopolysaccharide had no effect, suggesting that general inflammatory signals hold no sway over KLK7, but glutamate, long recognized as a potential excitotoxin in neurodegenerative conditions, suppressed the protease, said Tomita. So too did NMDA, the glutamate receptor agonist. In fact, Tomita reported that the NMDA antagonist memantine, which is approved for treating mild to moderate AD, increased astrocyte production of KLK7 eightfold.
Researchers at the meeting called Tomita’s data impressive. Some wanted to know if KLK7 degraded just Aβ monomers. Tomita said so far he has only tested Aβ from 7PA2 cells developed by Dominic Walsh, which release monomers and oligomers (Walsh et al., 2002). Daugherty had also reported that treating oligomers with KLK7 prevented their toxicity. Others wanted to know if knocking out KLK7 protected the APP mice from neurodegeneration. Tomita said that work is ongoing, but the animals are too young to assess. Konrad Beyreuther, University of Heidelberg, asked why memantine does not reduce amyloid in AD patients if it so strongly increases levels of the protease. Tomita said he thinks the drug is given too late to see an effect. He suggested that early in AD astrocytes become fatigued and stop producing the protease.
Shifting the focus to Aβ production, Lucía Chávez-Gutiérrez from VIB/KU Leuven, and Harald Steiner from Ludwig-Maximilians-University, Munich, shared new data on γ-secretase function. Chávez-Gutiérrez offered an explanation for why so many different familial AD mutations increase the production of Aβ42 over Aβ40. In APP the most aggressive mutations are in the transmembrane domain that interacts with presenilin, but others are scattered throughout the protein, suggesting the global structure helps dictate pathogenicity. Likewise, in presenilin 1, FAD mutations are scattered throughout the protein, meaning most of them cannot affect the active site of the enzyme directly. Instead, Chávez-Gutiérrez proposed that they weaken the stability of the enzyme-substrate complex. To test this, she used a simple trick. She raised the temperature of the enzyme assay on a hunch that mutant enzyme-substrate complexes would more readily dissociate as the temperature rose than wild-type complexes would. This would limit processivity—that is, γ-secretase’s ability to chop amino acids off the C-terminal end of the APP transmembrane domain. Michael Wolfe at Brigham and Women’s Hospital, Boston, had previously reported that γ-secretase made more of the longer Aβ42/Aβ43 peptides as the temperature heated up, but hadn’t looked at how FAD mutations affected this (Quintero-Monzon et al., 2011).
Chávez-Gutiérrez found that on turning up the heat to around 45 degrees Celsius, ε cleavage was hardly affected. But processivity fell, resulting in production of longer Aβ peptides, including Aβ43. She saw the same pattern for presenilin mutants; however, the shift toward longer peptides occurred at lower temperatures. Instability of the enzyme-substrate complex and longer Aβ peptide production correlated with a mutation’s pathogenicity, as judged by its expected age at disease onset. De Strooper, now at the U.K. Dementia Research Institute at University College London, considers the findings important. “This shows us that destabilization of presenilin lies at the heart of the pathogenesis,” he said. “Finally, we have a mechanism [for presenilin mutations] that translates into a clinical outcome.” Haass, from the German Center for Neurodegenerative Diseases in Munich, agreed. “The data are super nice and show how FAD mutations have subtle effects on protein structure,” he told Alzforum. He noted that it fits well with Steiner’s work.
For his part, Steiner remained unconvinced that weakened enzyme-substrate stability fully explains pathogenicity. “There is definitely a correlation that fits and is consistent, but whether it is generally the case, I’m not so sure,” he told Alzforum. He noted, for example, that there are also mutations within the N-terminal and mid-Aβ domain that increase the aggregation propensity of the peptide. He also found it puzzling that temperature didn’t affect ε cleavage as much as processivity.
Chávez-Gutiérrez thinks that’s because the mutations do not alter the affinity of APP substrates for γ-secretase but do promote dissociation, thereby reducing processivity. Others had shown that presenilin mutations enhance dissociation, aka off rate, of Aβ42 (Okochi et al., 2013). Chávez-Gutiérrez said something must be happening after the formation of the enzyme-substrate complex and before cleavage. “A potential scenario is that mutations, and temperature, have an impact on the stabilization of the transition state, which involves structural rearrangements on both enzyme and substrate. In some cases, those structural changes could lead to dissociation instead of catalysis,” she said.
Steiner thought this was probably the case. This could explain how certain mutants, such as PS1 M139V, generate longer Aβ peptides than wild-type presenilin but have normal endopeptidase (ε cleavage) activity (Chávez-Gutiérrez et al., 2012). Wolfe had also reported that activity, i.e., ε cleavage, and processivity can be dissociated by changing not only temperature but pH and salt concentration.
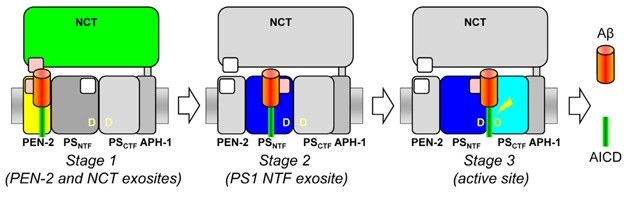
Three Steps to Aβ. APP substrates bind the γ-secretase complex in step-wise fashion, latching onto nicastrin/pen-2 first (left), before sidling onto an exosite on presenilin (middle), and then finally finding the active site (right). [Courtesy of Harald Steiner.]
In fact, substrates initially bind to γ-secretase in an exquisite choreography involving three of the complex’s subunits, as previously detailed by researchers in Steiner’s lab. Incorporating photo-labile amino acids into various positions in APP, they mapped its binding to γ-secretase. The C99 APP fragment generated by β-secretases binds first to exosites distinct from the catalytic site (Fukumori and Steiner, 2016). The substrate latches on first to pen-2/nicastrin, then to the N-terminal of presenilin before finally nestling in at the active site (see image above).
Does C83, the product of α-secretase cleavage of APP, bind to γ-secretase exosites in the same way, or even bypass them, given that its ectodomain is so short? Again, Steiner and colleagues used photo-affinity tags to map C83 binding to γ-secretase. While they found that it, too, bound to exosites before moving to the active site, the amino acids involved in binding were not the same. “We might be able to exploit this difference to develop a therapeutic that selectively blocks binding of C99 to γ-secretase,” said Steiner. He showed how this might work in principle. He could block binding and subsequent cleavage of C99 by mutating residues in the ectodomain that are not found in the shorter C83. “Conceptually, by targeting exosite-binding of C99, we could get a drug that spares C83 and Notch,” he suggested.
A Receptor for sAPP?
Turning to the other end of APP, De Strooper reported that Heather Rice, a postdoc in his lab, and Joris De Wit at KU Leuven, may have finally fingered a receptor for sAPP, the soluble ectodomain shed by β-secretase. Researchers have long hypothesized that this domain might trigger some type of cell signaling but a strong receptor candidate has not emerged. Given that APP concentrates in synapses, De Strooper and colleagues used a proteomic approach to screen for sAPP binding partners there and nabbed GABAb1 receptors as having high affinity for the ectodomain.
De Strooper explained how they narrowed down the interaction to GABAb1a, one of two isoforms of the receptor. Only GABAb1a contains so-called sushi domains, and the first of these turns out to be required for sAPP binding, De Strooper said. Sushi domains appear to traffic GABAb receptors to axons (Biermann et al., 2010). A 17 amino acid motif on the extension domain of sAPP appears to recognize the sushi domain.
Does sAPPb regulate the GABA receptor? Adding the fragment to hippocampal neurons reduced mini excitatory postsynaptic potentials. It had the same effect on CA3-CA1 circuits in hippocampal slices, De Strooper said. The findings suggest that sAPP has a positive effect on GABAb receptors to reduce synaptic transmission downstream.
Steiner deems this an interesting development and others agreed. Haass called it “remarkable,” noting that people have been looking for an sAPP receptor for many years in what has proven to be an extremely difficult search. “I consider this a really important finding,” he said.—Tom Fagan
References
Antibody Citations
Therapeutics Citations
Paper Citations
- Shropshire TD, Reifert J, Rajagopalan S, Baker D, Feinstein SC, Daugherty PS. Amyloid β peptide cleavage by kallikrein 7 attenuates fibril growth and rescues neurons from Aβ mediated toxicity in vitro. Biol Chem. 2013 Aug 30; PubMed.
- Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC. Single App knock-in mouse models of Alzheimer's disease. Nat Neurosci. 2014 May;17(5):661-3. Epub 2014 Apr 13 PubMed.
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002 Apr 4;416(6880):535-9. PubMed.
- Quintero-Monzon O, Martin MM, Fernandez MA, Cappello CA, Krzysiak AJ, Osenkowski P, Wolfe MS. Dissociation between the processivity and total activity of γ-secretase: implications for the mechanism of Alzheimer's disease-causing presenilin mutations. Biochemistry. 2011 Oct 25;50(42):9023-35. Epub 2011 Sep 30 PubMed.
- Okochi M, Tagami S, Yanagida K, Takami M, Kodama TS, Mori K, Nakayama T, Ihara Y, Takeda M. γ-secretase modulators and presenilin 1 mutants act differently on presenilin/γ-secretase function to cleave Aβ42 and Aβ43. Cell Rep. 2013 Jan 31;3(1):42-51. PubMed.
- Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261-74. Epub 2012 Apr 13 PubMed.
- Fukumori A, Steiner H. Substrate recruitment of γ-secretase and mechanism of clinical presenilin mutations revealed by photoaffinity mapping. EMBO J. 2016 Aug 1;35(15):1628-43. Epub 2016 May 23 PubMed.
- Biermann B, Ivankova-Susankova K, Bradaia A, Abdel Aziz S, Besseyrias V, Kapfhammer JP, Missler M, Gassmann M, Bettler B. The Sushi domains of GABAB receptors function as axonal targeting signals. J Neurosci. 2010 Jan 27;30(4):1385-94. PubMed.
Further Reading
Papers
- Ito K, Tatebe T, Suzuki K, Hirayama T, Hayakawa M, Kubo H, Tomita T, Makino M. Memantine reduces the production of amyloid-β peptides through modulation of amyloid precursor protein trafficking. Eur J Pharmacol. 2017 Mar 5;798:16-25. Epub 2017 Feb 4 PubMed.
- Quintero-Monzon O, Martin MM, Fernandez MA, Cappello CA, Krzysiak AJ, Osenkowski P, Wolfe MS. Dissociation between the processivity and total activity of γ-secretase: implications for the mechanism of Alzheimer's disease-causing presenilin mutations. Biochemistry. 2011 Oct 25;50(42):9023-35. Epub 2011 Sep 30 PubMed.
- Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chávez-Gutiérrez L. Alzheimer's-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell. 2017 Jul 27;170(3):443-456.e14. PubMed. Correction.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.