TREM2 Data Surprise at SfN Annual Meeting
Quick Links
Research into what microglia do in Alzheimer's disease got a boost when geneticists discovered that mutations in the glial cell surface receptor TREM2 increase the risk for AD. But is the problem really with the microglia? At the Society for Neuroscience annual meeting, held November 15 to 19 in Washington, D.C., researchers surprised the audience by claiming that TREM2-expressing cells in the vicinity of Aβ plaques in the brain are monocytes that came in from the blood. Even more surprising, the new research links reduced TREM2 to attenuation of amyloid plaque load in the hippocampus. "This could totally change our assumptions about Aβ clearance," said Gary Landreth, Case Western Reserve University, Cleveland, who directed the work along with Bruce Lamb at the Cleveland Clinic. Scientists who saw the presentations suggested cautious interpretation, noting previous studies suggesting that microglia make more TREM2 than peripheral macrophages do and that a loss of TREM2 function puts people at risk for neurodegenerative diseases, including Alzheimer’s.
The basis for the peripheral monocyte claim comes from flow cytometry analysis of cells expressing TREM2. Crystal Miller, from Lamb's lab, examined TREM2 in 5xFAD and APP/PS1 mouse models of AD, both of which aggressively develop amyloid plaques. She reported that as the mice aged, TREM2 increased in the brain and co-localized with Congo Red-positive Aβ plaques as judged by immunohistochemistry with a TREM2 antibody. She crossed a mouse with a Lac Z gene knocked into the TREM2 locus with the AD models and, when she looked at amyloid plaques in their brains, saw the characteristic blue staining that this bacterial enzyme generates in response to the sugar compound X-gal. This helped confirm that glia around plaques express TREM2. Miller told Alzforum that questions had been raised about the specificity of TREM2 antibodies and she wanted to ensure she was looking at TREM2, not some other epitope. When she knocked out the TREM2 gene she saw no expression of the protein, confirming the antibody’s specificity. Using immunohistochemistry, Miller also saw TREM2-positive cells around Aβ deposits in human brain tissue.
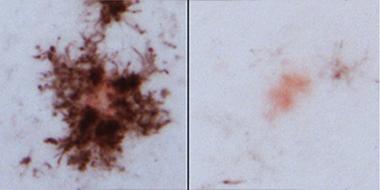
Monocytes surround plaques:
In APP/PS1 mice, CD45-positive cells (dark brown) surround Congo Red-positive plaques (left), but not if TREM2 is knocked out (right). [Image courtesy of Crystal Miller.]
What are the TREM2-positive cells crowding around amyloid plaques? They lacked the purinergic receptor P2RY12, a marker of microglia. Instead, they flaunted Iba1, a marker of pro-inflammatory, non-phagocytic cells. This contravenes dogma, Landreth told Alzforum. "The literature suggests that TREM2 is anti-inflammatory and pro-phagocytotic, but here we found plaques associating with TREM2-positive cells that must be pro-inflammatory and phagocytically inactive based on the vast Iba1 literature," he said.
To characterize the TREM2 cells further, Miller isolated them by flow cytometry, then analyzed their other markers. To her surprise, she found TREM2 in cells that highly expressed CD45. "This was a huge shock," said Miller. Scientists believe that cells flush with CD45 are peripheral monocytes, not brain-resident microglia. The TREM2-positive cells expressed other markers of peripheral cells as well, including F480 and Ly6C.
To Miller, the data suggest that TREM2-expressing cells in the brain must be recruited from the periphery. In support of this, she found four- to fivefold more TREM2-positive cells in the blood of 4-month-old APPPS1 mice than in the blood of wild-type controls. While this suggests that peripheral monocytes in AD mouse models make TREM2 while in the blood, Miller said that the cells might also begin to express the protein after they have infiltrated the brain.
Researchers at the meeting seemed puzzled by the findings. Dave Morgan, University of South Florida, Tampa, cautioned that Joseph El Khoury at Massachusetts General Hospital, Charlestown, and others have shown that microglia preferentially express TREM2 (see Thrash et al., 2009, and Hickman et al., 2013). At the same time, El Khoury's lab has implicated peripheral monocytes in clearing plaques (see Mar 2007 news story). "The microglial role just gets more interesting," said Morgan.
Others wondered if the findings might be specific to mice. Both Elliott Mufson from Rush University, Chicago, and Greg Cole of University of California, Los Angeles, noted that TREM2 expression ticks up only slightly in the AD brain, much less than in mouse models. Christian Haass, Ludwig-Maximilians-Universität, Munich, wondered if there might be something about these mice that prompts their peripheral cells to invade the brain. "Even how the animals are housed could be an important factor," he told Alzforum.
Monica Carson from the University of California, Riverside, who was not at the meeting but had heard about the findings, said the data get people to think about TREM2 biology. She said it was plausible that TREM2 cells might infiltrate the brain from the periphery. "We are starting to realize that the biology of TREM2 is not as simple as turning it on or off, or it being expressed in one cell rather than the other," she told Alzforum. Carson believes that the key to TREM2 expression in AD models is the milieu around plaques. "That environment might demand induction of TREM2 on whatever immune cell is there," she suggested. Carson previously reported that TREM2-positive cells around plaques are most likely brain-resident microglia (see Melchior et al., 2010). That was in the APP23 model, which lays down plaques more slowly than the 5xFAD and APP/PS1 models that Miller and colleagues used.
Carson also urged caution in interpreting TREM2 antibody interactions. Several groups have reported that TREM2’s ectodomain sheds from the cell surface and can bind to other nearby cells (see Hamerman et al., 2006, and Wunderlich et al., 2013). Antibodies that recognize that ectodomain may not distinguish between cells that express TREM2 and those that bind it, she said.
The origin of TREM2 cells aside, what effect does loss of the protein have on plaques? Taylor Jay, a graduate student in the labs of both Landreth and Lamb, reported that far fewer glia surrounded plaques in TREM2 knockout APP/PS1 mice than in control APP/PS1 animals. In fact, the even spacing, or tiling, of microglia seen in a normal mouse brain was about the same in APP/PS1 TREM2 knockouts, suggesting that TREM2 is essential for recruiting cells to plaques. "There are very few plaque-associated microglia in the TREM2 knockouts," said Landreth. This fits with work by Jason Ulrich and colleagues at Washington University, St. Louis, who found fewer glia surrounding plaques in APP/PS1 animals that have only one copy of the TREM2 gene (see Jun 2014 news story). Jay also reported fewer astrocytes around plaques in APP/PS1 TREM2 knockouts compared with APP/PS1 controls. Inflammation was down as well, judging by reductions in pro-inflammatory interleukin 1b and IL6.
Corroborating Ulrich's finding, Jay found as many amyloid plaques in the cerebral cortex of the APP/PS1 TREM2 knockouts as in controls. Ulrich has since extended his observations to homozygous knockouts, and reported at SfN that they resemble the heterozygotes. In the latter, glial numbers were down by half, while in the homozygote knockouts they fell further, to 25 percent of controls. He said cortical plaque load was unchanged in either case.
Unexpectedly, while Jay found no change in plaque load in the cortex, she reported fewer plaques in the hippocampus in her APP/PS1/TREM2-/- mice. Immunohistochemistry using the 6E10 anti-Aβ antibody suggested more than a threefold reduction in plaques in 4-month-old animals. ELISA of brain lysates showed almost a twofold reduction in insoluble Aβ, as well. The tau antibodies AT8 and AT180 detected twofold less phospho-tau in the vicinity of plaques in the TREM2 knockouts. "We found these results very surprising based on predicted TREM2 function," said Jay. "If anything, we would have expected to see an increase in pathology in TREM2 knockouts."
Carson was not surprised by the drop in plaques. "That would be consistent with models of stroke, where we see decreased pathology in TREM2 knockouts," she said (see Sieber et al., 2013). She noted that TREM2 expression in the mouse brain is very high during early development, but falls considerably by the time the animals reach 3 months of age. "That suggests there is some risk to having high expression," she said.
The overall feeling at this meeting was that much more work needs to be done to understand the role of TREM2 in disease. Some scientists wondered if TREM2-positive cells were really coming from the periphery, or if brain-resident microglia were changing their phenotype to express markers typically considered peripheral. Others noted that any model for TREM2 function needs to explain not just pathology of AD, but of FTD, PD, and other neurodegenerative diseases genetically linked to TREM2, and suggested that clearance of dead and dying cells by microglia might be key. Haass, who presented his recent findings linking TREM2 mutations to compromised microglial phagocytosis, agreed (see Kleinberger et al., 2014, and Alzforum webinar). "We see that removal of debris and cells is abolished in TREM2 knockouts and this may be the best way to look at its function," he said. —Tom Fagan
References
Research Models Citations
News Citations
Webinar Citations
Paper Citations
- Thrash JC, Torbett BE, Carson MJ. Developmental regulation of TREM2 and DAP12 expression in the murine CNS: implications for Nasu-Hakola disease. Neurochem Res. 2009 Jan;34(1):38-45. Epub 2008 Apr 11 PubMed.
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013 Dec;16(12):1896-905. Epub 2013 Oct 27 PubMed.
- Melchior B, Garcia AE, Hsiung BK, Lo KM, Doose JM, Thrash JC, Stalder AK, Staufenbiel M, Neumann H, Carson MJ. Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine-based therapies for Alzheimer's disease. ASN Neuro. 2010 Jul 12;2(3):e00037. PubMed.
- Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol. 2006 Aug 15;177(4):2051-5. PubMed.
- Wunderlich P, Glebov K, Kemmerling N, Tien NT, Neumann H, Walter J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) by ectodomain shedding and γ-secretase dependent intramembranous cleavage. J Biol Chem. 2013 Nov 15;288(46):33027-36. PubMed.
- Sieber MW, Jaenisch N, Brehm M, Guenther M, Linnartz-Gerlach B, Neumann H, Witte OW, Frahm C. Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS One. 2013;8(1):e52982. Epub 2013 Jan 3 PubMed.
- Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014 Jul 2;6(243):243ra86. PubMed.
Further Reading
Papers
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL, Cotleur AC, Butovsky O, Bekris L, Staugaitis SM, Leverenz JB, Pimplikar SW, Landreth GE, Howell GR, Ransohoff RM, Lamb BT. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J Exp Med. 2015 Mar 9;212(3):287-95. Epub 2015 Mar 2 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.