With TREM2, Timing Is Everything
Quick Links
Certain variants in the TREM2 gene more than triple a person’s risk of AD, seemingly by sapping the protective function this microglial receptor performs. According to findings presented at the virtual AAT-AD/PD meeting, held April 2 to 5, TREM2’s protective power falters as amyloidosis kicks into high gear.
- 5xFAD mice expressed human TREM2 at different ages.
- WT TREM2 slowed plaques early but not later on.
- R47H-TREM2 never protected, and worsened amyloid deposition during seeding stage.
Guojun Bu of the Mayo Clinic in Jacksonville, Florida, described what happened in a mouse model of amyloidosis when he switched on expression of human TREM2 at different stages of disease. Wild-type human TREM2 stemmed Aβ deposition, but only while plaques were in their infancy. It had no effect later on. In contrast, the R47H mutant never protected against plaques, and even exacerbated their growth. Together, the findings suggest that TREM2’s protective role is limited to early stages of Aβ deposition.
The heightened AD risk posed by rare variants in TREM2 point to a beneficial role for the wild-type variant, yet animal models have painted a complex and sometimes contradictory picture of the receptor’s influence on AD pathogenesis. For one, expression of TREM2 on microglia ramps up as amyloidosis worsens, yet microglia ultimately morph into ineffective, or even harmful, pro-inflammatory cells (Jan 2017 news; Sep 2017 news; Jun 2018 conference news).
Soluble TREM2, which is cleaved from the microglial surface, helps recruit microglia to plaques, yet levels of the snipped receptor rise in human cerebrospinal fluid as cognitive symptoms emerge in people with AD (Apr 2019 news; Jan 2016 news).
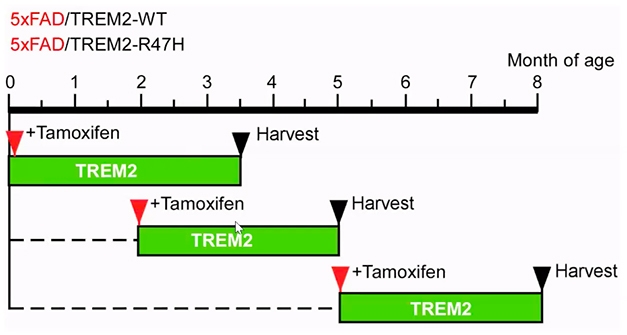
Flipping the TREM2 Switch. Tamoxifen induced expression of human TREM2 in 5xFAD mice at different stages of amyloidosis. [Courtesy of Guojun Bu, Mayo Clinic.]
How does TREM2’s role change throughout the long, drawn-out stages of amyloidosis? To address this question, Bu developed inducible human TREM2 mouse strains. Treating the mice with tamoxifen switched on expression of either human wild-type TREM2 or the R47H variant in microglia, while endogenous mouse TREM2 expression stayed constant. The researchers then crossed these TREM2 mice to the 5xFAD mouse model of amyloidosis. In the crosses, they induced human TREM2 expression during three phases of amyloidosis: early seeding and growth (birth to 3.5 months), rapid growth (2 to 5 months), or maturation (5 to 8 months) (see above).
When expressed from birth to 3.5 months, human wild-type TREM2 strongly protected against plaque formation. Mice expressing human wild-type TREM2 had about fourfold fewer plaques than did non-induced controls. In contrast, expression of R47H-TREM2 had no effect on plaque deposition at this early stage.
Everything changed when human TREM2 expression was on from 2 to 5 months of age. At this slightly older age, wild-type TREM2 expression no longer slowed amyloid deposition, and R47H made matters worse. Animals expressing the risk variant had two to three times more plaques than non-induced controls, though the plaques were about the same size in both strains.
The data suggest that during this phase of rapid plaque growth, wild-type TREM2 loses its protective function, while the R47H variant appears to exert a toxic function. In support of this idea, single-cell transcriptomic experiments revealed that the R47H mutant invoked more dramatic shifts in the microglial transcriptome than did the wild-type allele. In 5-month-old mice, R47H-TREM2 but not wild-type TREM2 upregulated genes involved in dysfunctional cell stress and energy metabolism pathways in microglia.
When expressed during the maturation phase of beta amyloidosis, from 5 to 8 months of age in these mice, neither wild-type nor mutant TREM2 affected Aβ deposition.
Together, the findings suggest that normal TREM2 thwarts amyloidosis early on, when plaques are just starting to develop. Once plaque growth takes off, TREM2’s protective function fades, and the detrimental effects of R47H become apparent. By the maturation phase, neither version of the human protein holds sway over amyloid.
The findings could have implications for the timing of therapeutic strategies aimed at revving up TREM2 signaling, Bu said. Such strategies would be most likely to work early in disease.—Jessica Shugart
References
Mutations Citations
News Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.