Alzheimer’s Gene BIN1 Promotes Synaptic Transmission
Quick Links
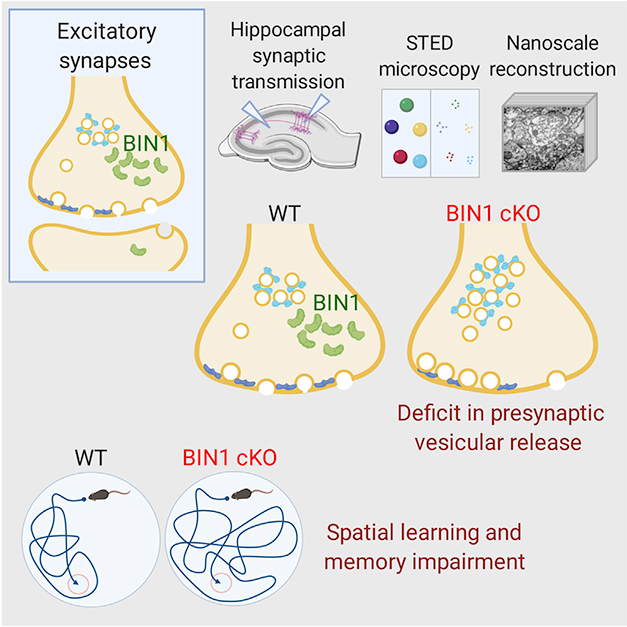
BIN1, one of the strongest genetic risk factors for late-onset Alzheimer’s disease, encodes a protein that dabbles in many cellular functions, taking a hand in Aβ production, tau propagation, and microglial function. Now, in the March 10 Cell Reports, researchers led by Gopal Thinakaran at the USF Morsani College of Medicine in Tampa, Florida, tie it to synaptic transmission, as well. At least in mice. The BIN1 protein clustered with presynaptic vesicles at excitatory nerve terminals, the authors found. Neurons lacking the protein were less likely to release these vesicles upon electrical stimulation. Their reticence dampened neuronal signaling and impaired spatial memory. Thinakaran previously discussed the data at the 2019 AD/PD conference (May 2019 conference news).
- In mice, loss of neuronal BIN1 weakens excitatory signaling.
- Without BIN1, the hippocampus had fewer synapses …
- … and the mice couldn’t learn spatial information.
“These results raise the intriguing possibility that presynaptic BIN1 alterations may contribute to memory impairment [in Alzheimer’s disease],” Michael Ewers at Ludwig-Maximilians-University in Munich wrote to Alzforum (full comment below). He was not involved in the research.
BIN1 is an adaptor protein known to affect cell membrane dynamics such as endocytosis and exocytosis. Previously, researchers linked BIN1 to both amyloid and tau pathology (Apr 2015 conference news; Oct 2016 news; Apr 2019 conference news). Other research has placed BIN1 in the postsynapse (Nov 2015 conference news).
Vesicle Buildup. Without BIN1 (green), vesicles pile up in excitatory presynaptic nerve terminals (right), compared with wild-types (left). This causes memory loss. [Courtesy of De Rossi et al., Cell Reports.]
Thinakaran and colleagues explored the synaptic effects of conditionally knocking out BIN1 in neurons. First author Pierre de Rossi generated mice that lost the gene in cells with an active Syn1 promoter, i.e., primarily neurons in the hippocampus and to a lesser extent in the cortex. The authors prepared hippocampal slice cultures from these BIN1 knockout mice and wild-type controls. Knockout neurons were less likely to release synaptic vesicles upon stimulation than were controls. Curiously, however, they maintained synaptic transmission longer than did controls during sustained stimulation. Together, the results suggest Bin1 knockout neurons keep a larger pool of synaptic vesicles in reserve.
Electron microscopy bore out these findings. Axon terminals of knockout neurons contained more docked vesicles, and a larger reserve pool of vesicles, than controls. Super-resolution STED microscopy revealed alterations in several presynaptic proteins, with the density of synaptoporin, synaptotagmin-1, and Munc13-1 down, and synaptophysin, synaptojanin, and Rab5 up. The net effect of these changes would be to retard vesicle release, Thinakaran noted.
In addition to these presynaptic changes, Bin1 knockouts had fewer hippocampal synapses than controls. In the latter, most BIN1 loitered in axons, where it associated with clusters of synaptic vesicles. In excitatory neurons, 21 percent of presynapses and 6 percent of postsynapses contained the protein (see image below).
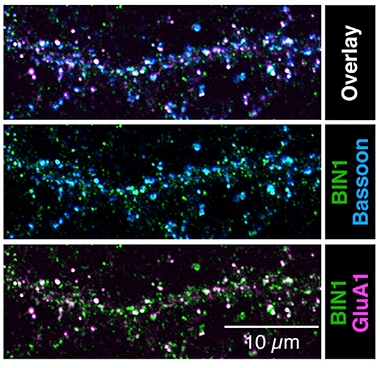
BIN1 Favors Presynapses. In cultured wild-type hippocampal neurons, BIN1 (green) frequently occurs together with the presynaptic marker Bassoon (blue, overlay appears aqua, top), but rarely with postsynaptic marker GluA1 (purple). [Courtesy of De Rossi et al., Cell Reports.]
Behavioral testing supported the idea of neurotransmission deficits. Knockout mice were unable to learn the location of a hidden platform in the Morris water maze, a test of hippocampal spatial memory. Other frequently tested forms of mouse learning, however, including fear conditioning, novel object recognition, and the Y maze, appeared normal.
The authors tested a second conditional BIN1 knockout model using a broader promoter, Emx1. It excised the gene in hippocampal and in cortical excitatory neurons, and also in glia. This model had similar alterations to presynaptic proteins as Syn1-BIN1 knockouts did. The Emx1-BIN1 knockouts fared poorly in the Morris water maze, and struggled to remember objects they had seen before. This additional memory impairment might be a result of BIN1 loss in forebrain neurons, which participate in recognition memory, the authors suggested.
Gunnar Gouras at Lund University, Sweden, praised the use of several types of high-resolution microscopy along with behavioral data in two knockout models. “This is an excellent study that provides novel and convincing evidence for BIN1 acting in regulating release probability at presynapses,” he wrote to Alzforum (full comment below).
Notably, BIN1 levels are abnormal, and mostly lower, in AD brain (Holler et al., 2014; De Rossi et al., 2016; McKenzie et al., 2017). “Together, the results from our conditional knockout study and the human AD data suggest that the loss of neuronal BIN1 function in synaptic physiology likely contributes to cognitive decline in AD,” Thinakaran wrote to Alzforum.
Thinakaran and colleagues did not look at whether any of the BIN1 AD risk variants affected synaptic transmission. A recent study by Christopher Glass and colleagues at the University of California, San Diego, found that the main risk variant affects expression in microglia only (Jul 2018 conference news; Nov 2019 news). However, Glass told Alzforum that this finding does not rule out a contribution from other cell types, because transcription may change in the AD brain (full comment below).
Ewers agreed. “The current study adds to a heterogenous mosaic of BIN1 functions and their association with cognitive decline and AD dementia risk … Taken together, BIN1 related patho-mechanisms in AD are complex, and future studies combining preclinical research and investigations in AD patients will be needed,” he wrote.—Madolyn Bowman Rogers
References
News Citations
- Off-Balance Endocytosis Lays Groundwork for Disease
- The Feud, Act II: Do Alzheimer’s Genes Affect Amyloid or Tau?
- Lack of BIN1 Sows Tau Trouble for Neurons
- Parsing How Alzheimer’s Genetic Risk Works Through Microglia
- Alzheimer’s GWAS Hits Point to Endosomes, Synapses
- A Delicate Frontier: Human Microglia Focus of Attention at Keystone
- Cell-Specific Enhancer Atlas Centers AD Risk in Microglia. Again.
Paper Citations
- Holler CJ, Davis PR, Beckett TL, Platt TL, Webb RL, Head E, Murphy MP. Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer's disease brain and correlates with neurofibrillary tangle pathology. J Alzheimers Dis. 2014;42(4):1221-7. PubMed.
- De Rossi P, Buggia-Prévot V, Clayton BL, Vasquez JB, van Sanford C, Andrew RJ, Lesnick R, Botté A, Deyts C, Salem S, Rao E, Rice RC, Parent A, Kar S, Popko B, Pytel P, Estus S, Thinakaran G. Predominant expression of Alzheimer's disease-associated BIN1 in mature oligodendrocytes and localization to white matter tracts. Mol Neurodegener. 2016 Aug 3;11(1):59. PubMed. Correction.
- McKenzie AT, Moyon S, Wang M, Katsyv I, Song WM, Zhou X, Dammer EB, Duong DM, Aaker J, Zhao Y, Beckmann N, Wang P, Zhu J, Lah JJ, Seyfried NT, Levey AI, Katsel P, Haroutunian V, Schadt EE, Popko B, Casaccia P, Zhang B. Multiscale network modeling of oligodendrocytes reveals molecular components of myelin dysregulation in Alzheimer's disease. Mol Neurodegener. 2017 Nov 6;12(1):82. PubMed.
Further Reading
Primary Papers
- De Rossi P, Nomura T, Andrew RJ, Masse NY, Sampathkumar V, Musial TF, Sudwarts A, Recupero AJ, Le Metayer T, Hansen MT, Shim HN, Krause SV, Freedman DJ, Bindokas VP, Kasthuri N, Nicholson DA, Contractor A, Thinakaran G. Neuronal BIN1 Regulates Presynaptic Neurotransmitter Release and Memory Consolidation. Cell Rep. 2020 Mar 10;30(10):3520-3535.e7. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Ludwig Maximilian University
Ludwig Maximilians University
Genetic variants near BIN1 are important risk factors for AD dementia, where the SNP rs744373 is the second most strongly associated genetic locus for sporadic AD after the ApoE ε4 allele. Despite these strong genetic associations of BIN1 with AD dementia, the patho-mechanisms that link BIN1 alterations to AD remain unclear. We and others have previously shown that genetic variants of BIN1 are selectively associated with increased tau pathology rather than Aβ pathology (Chapuis et al., 2013; Franzmeier et al., 2019), which may explain increased AD dementia risk in BIN1 SNP carriers. BIN1 is part of a larger network of AD genetic risk variants that encode proteins involved in endocytosis and vesicle-mediated trafficking (Schurmann et al., 2019). A recent study by Calafate and colleagues showed that BIN1 genetic variants increase the endocytosis of tau and may thus enhance its spread (Calafate et al., 2015). However, distinct from its involvement in postsynaptic endocytosis, BIN1 has several other functions, such as postsynaptic recycling, exocytosis, and exosome trafficking, that may contribute to increased tau pathology when pathologically altered (Crotti et al., 2019; Schurmann et al., 2019).
This study by de Rossi et al. draws attention to presynaptic function of BIN1, showing that it localizes to presynaptic sites, particularly in excitatory neurons, where BIN1 knockout in mice leads to reduced vesicle release, low synaptic density, and reduced spatial memory performance. These results raise the intriguing possibility of presynaptic BIN1 alterations that may contribute to memory impairment. An important open question is whether presynaptic BIN1-related alterations may contribute to genetic AD dementia risk conferred by BIN1 variants through increasing tau pathology.
The current study adds to a heterogenous mosaic of BIN1 functions and their association with cognitive decline and AD dementia risk. Recent studies suggest yet another potential role of BIN1, i.e., regulation of microglia activation and thus changes in tau pathology (Crotti et al., 2019; Nott et al., 2019). Taken together, BIN1 related patho-mechanisms in AD are complex, and future studies, combining preclinical research and investigations in AD patients, will be needed to identify those mechanism that drive disease progression in AD and to work toward a potential therapeutic application.
References:
Calafate S, Buist A, Miskiewicz K, Vijayan V, Daneels G, de Strooper B, de Wit J, Verstreken P, Moechars D. Synaptic Contacts Enhance Cell-to-Cell Tau Pathology Propagation. Cell Rep. 2015 May 26;11(8):1176-83. Epub 2015 May 14 PubMed.
Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwenberghe C, Kolen KV, Geller F, Sottejeau Y, Harold D, Dourlen P, Grenier-Boley B, Kamatani Y, Delepine B, Demiautte F, Zelenika D, Zommer N, Hamdane M, Bellenguez C, Dartigues JF, Hauw JJ, Letronne F, Ayral AM, Sleegers K, Schellens A, Broeck LV, Engelborghs S, De Deyn PP, Vandenberghe R, O'Donovan M, Owen M, Epelbaum J, Mercken M, Karran E, Bantscheff M, Drewes G, Joberty G, Campion D, Octave JN, Berr C, Lathrop M, Callaerts P, Mann D, Williams J, Buée L, Dewachter I, Van Broeckhoven C, Amouyel P, Moechars D, Dermaut B, Lambert JC, GERAD consortium. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry. 2013 Nov;18(11):1225-34. Epub 2013 Feb 12 PubMed.
Crotti A, Sait HR, McAvoy KM, Estrada K, Ergun A, Szak S, Marsh G, Jandreski L, Peterson M, Reynolds TL, Dalkilic-Liddle I, Cameron A, Cahir-McFarland E, Ransohoff RM. BIN1 favors the spreading of Tau via extracellular vesicles. Sci Rep. 2019 Jul 1;9(1):9477. PubMed.
Franzmeier N, Rubinski A, Neitzel J, Ewers M, Alzheimer’s Disease Neuroimaging Initiative (ADNI). The BIN1 rs744373 SNP is associated with increased tau-PET levels and impaired memory. Nat Commun. 2019 Apr 16;10(1):1766. PubMed.
Nott A, Holtman IR, Coufal NG, Schlachetzki JC, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, Keulen Z, Pasillas MP, O'Connor C, Nickl CK, Schafer ST, Shen Z, Rissman RA, Brewer JB, Gosselin D, Gonda DD, Levy ML, Rosenfeld MG, McVicker G, Gage FH, Ren B, Glass CK. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science. 2019 Nov 29;366(6469):1134-1139. Epub 2019 Nov 14 PubMed.
Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013 Apr;14(4):389-94. PubMed.
Schürmann B, Bermingham DP, Kopeikina KJ, Myczek K, Yoon S, Horan KE, Kelly CJ, Martin-de-Saavedra MD, Forrest MP, Fawcett-Patel JM, Smith KR, Gao R, Bach A, Burette AC, Rappoport JZ, Weinberg RJ, Martina M, Penzes P. A novel role for the late-onset Alzheimer's disease (LOAD)-associated protein Bin1 in regulating postsynaptic trafficking and glutamatergic signaling. Mol Psychiatry. 2019 Apr 9; PubMed.
Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RA, Herman M, Nahmani E, Emrani S, Figueroa YH, Diamond MI, Clelland CL, Wray S, Duff KE. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 2016 Aug;19(8):1085-92. Epub 2016 Jun 20 PubMed.
Lund University
This is an excellent study that provides novel and convincing evidence for BIN1 acting in regulating release probability at presynapses. Synapses are increasingly seen as the vulnerable early targets in AD, and determining molecular mechanisms by which genetic risk factors affect AD is of major importance. The combination of two different conditional Bin1 cKO mice with behavioral studies, electrophysiology, and super-resolution and immuno-EM of Bin1 localization and synaptic alterations is high-level work.
De Rossi and colleagues mention in their Introduction that their prior paper showed that Bin1 KO mice crossed with AD transgenics did not alter “beta-amyloid pathogenesis” and also discuss how other published work differs from their current results (Andrew et al., 2019). Having focused on synapses in AD for years, I can suggest reasons for why BIN1 as a risk factor of AD does not affect plaque pathology but still might affect Aβ and why results might differ between groups when studying neurons.
The authors cite papers on greater effects of BIN1 on tau than amyloid pathologies; however, they could consider more papers showing preplaque synapse dysfunction. Increasing evidence points to Aβ oligomers, and while the field is quite aware of this, there seems continued surprise when plaques don’t correlate with impairment. If BIN1 is an important AD risk factor and Aβ is tightly linked to AD, then how might one explain no change in plaques with Bin1 KO? Recall, plaques do not correlate with cognition in human AD. For example, some Aβ antibodies can remove plaques without improving cognition, while other antibodies can improve cognition without removing plaques. Thus, unaltered plaques in Bin1 KO mice, does not have to mean that BIN1 is “Aβ -independent,” a popular term the past few years.
Functioning synapses are important to generate plaques and impairing synapse function can lead to less plaques but also more synapse damage. We showed that altering activity in AD transgenic mouse brains led to less plaques but more loss of synapses (Tampellini et al., 2010). Thus, AD transgenic mice crossed with Bin1 KO mice might be re-examined for: 1. reduction in synapse proteins, such as synaptophysin (expected as per the current De Rossi et al), and 2. for not just total Aβ (or plaques) but for Aβ accumulation specifically in synapses (high resolution imaging or use of synaptosomes; see e.g., Bilousova et al., 2019). The lack of behavioral impairment reported by Andrew et al., was important, although lack of behavioral change in mice, and even in humans, can occur with quite a lot of brain pathology.
In terms of their discussion of different results by different groups, here synapses can also be tricky, because they certainly are not all the same, and anatomy and neuron subtype can be critical. As one example, we saw APP C99 accumulation with PS1cKO particularly in presynapses of CA1 but postsynapses of CA3 (Willén et al., 2017).
Thus, this paper; Schürmann et al., 2019; Miyagawa et al., 2016; and Ubelmann et al., 2017, all provide exciting, albeit not the same, new clues to the cellular mechanisms in neurons by which BIN1 can impact AD.
References:
Andrew RJ, De Rossi P, Nguyen P, Kowalski HR, Recupero AJ, Guerbette T, Krause SV, Rice RC, Laury-Kleintop L, Wagner SL, Thinakaran G. Reduction of the expression of the late-onset Alzheimer's disease (AD) risk-factor BIN1 does not affect amyloid pathology in an AD mouse model. J Biol Chem. 2019 Mar 22;294(12):4477-4487. Epub 2019 Jan 28 PubMed.
Tampellini D, Capetillo-Zarate E, Dumont M, Huang Z, Yu F, Lin MT, Gouras GK. Effects of synaptic modulation on beta-amyloid, synaptophysin, and memory performance in Alzheimer's disease transgenic mice. J Neurosci. 2010 Oct 27;30(43):14299-304. PubMed.
Bilousova T, Melnik M, Miyoshi E, Gonzalez BL, Poon WW, Vinters HV, Miller CA, Corrada MM, Kawas C, Hatami A, Albay R 3rd, Glabe C, Gylys KH. Apolipoprotein E/Amyloid-β Complex Accumulates in Alzheimer Disease Cortical Synapses via Apolipoprotein E Receptors and Is Enhanced by APOE4. Am J Pathol. 2019 Aug;189(8):1621-1636. Epub 2019 May 17 PubMed.
Willén K, Edgar JR, Hasegawa T, Tanaka N, Futter CE, Gouras GK. Aβ accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Mol Neurodegener. 2017 Aug 23;12(1):61. PubMed.
Schürmann B, Bermingham DP, Kopeikina KJ, Myczek K, Yoon S, Horan KE, Kelly CJ, Martin-de-Saavedra MD, Forrest MP, Fawcett-Patel JM, Smith KR, Gao R, Bach A, Burette AC, Rappoport JZ, Weinberg RJ, Martina M, Penzes P. A novel role for the late-onset Alzheimer's disease (LOAD)-associated protein Bin1 in regulating postsynaptic trafficking and glutamatergic signaling. Mol Psychiatry. 2019 Apr 9; PubMed.
Miyagawa T, Ebinuma I, Morohashi Y, Hori Y, Young Chang M, Hattori H, Maehara T, Yokoshima S, Fukuyama T, Tsuji S, Iwatsubo T, Prendergast GC, Tomita T. BIN1 regulates BACE1 intracellular trafficking and amyloid-β production. Hum Mol Genet. 2016 May 14; PubMed.
Ubelmann F, Burrinha T, Salavessa L, Gomes R, Ferreira C, Moreno N, Guimas Almeida C. Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep. 2017 Jan;18(1):102-122. Epub 2016 Nov 28 PubMed.
University of California San Diego
The paper from De Rossi and colleagues reporting roles of neuronal BIN1 in regulation of presynaptic neurotransmitter release and memory consolidation is of substantial interest. BIN1 is expressed in neurons, microglia, and other non-neuronal cells within the brain, and may have different functions in each cell type. From our recent analysis of transcriptional enhancers in the major brain cell types derived from pediatric surgical samples, the most significant AD risk SNP associated with BIN1 resided in a microglia-specific enhancer (Nott et al., 2019; Nov 2019 news).
This observation suggests that the risk association is due to functions of BIN1 in microglia. However, this mechanism cannot be considered conclusive at this stage because the transcriptional landscapes of relevant cell types may be different in the context of AD and/or the causal variants may be associated with risk due to other mechanisms. An important question going forward with respect to the findings of De Rossi et al. is whether any of the noncoding AD risk SNPs associated with BIN1 alter its function in neurons of the brains of individuals with AD.
References:
Nott A, Holtman IR, Coufal NG, Schlachetzki JC, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, Keulen Z, Pasillas MP, O'Connor C, Nickl CK, Schafer ST, Shen Z, Rissman RA, Brewer JB, Gosselin D, Gonda DD, Levy ML, Rosenfeld MG, McVicker G, Gage FH, Ren B, Glass CK. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science. 2019 Nov 29;366(6469):1134-1139. Epub 2019 Nov 14 PubMed.
NOVA Medical School
De Rossi et al. address the physiological role of Bin1 in the brain, specifically at synapses. BIN1 is a susceptibility locus for late-onset Alzheimer’s disease (Seshadri et al., 2010); however, whether Bin1 is linked to the synapse dysfunction that best correlates with AD cognitive impairment is unclear (Tampellini and Gouras, 2010).
Using neuronal conditional knockout (cKO) mice models, the authors investigated Bin1's role in excitatory neurons. These models were generated by crossing the Bin1fl/fl strain with the Syn1-Cre, targeting hippocampal neurons, and Emx1-IRES-Cre driver lines, targeting excitatory neurons and glia. Both the neuronal and ubiquitous BIN1 isoforms are reduced in the brains of the two conditional KO mice studied, one KO of Bin1 in excitatory neurons (Emx) and the other in hippocampal cells (Syn).
In both models, the authors identify a role for BIN1 in excitatory synaptic transmission with an impact on learning and long-term memory consolidation. Mechanistically, a role in neurotransmitter vesicle release is proposed since Bin1 cKO mice show an accumulation of reserve pool and docked SV.
The authors did not observe significant alterations at the postsynaptic site, and suggest that the small changes perceived are compensatory of a presynaptic defect. A presynaptic function agrees with our data (Ubelmann et al., 2017), demonstrating that Bin1 polarizes toward axons, and with Bin1 presynaptic localization initially proposed by Di Paolo (2002). However, it contrasts with a recent paper from Peter Penzes, showing that Bin1 functions postsynaptically in the control of AMPA receptor trafficking and transmission (Schürmann et al., 2019).
How Bin1 controlled the number of vesicles in the reserve pool and docked at the plasma membrane remains to be understood. Given the known function of Bin1 in the scission of vesicles, it is not clear how the loss of scission could lead to an increase in SV number. The authors propose, instead, that Bin1 plays a role in SV release through the interaction with the SNARE complex. Nevertheless, this study provides one more piece of the puzzle of how BIN1 can impact LOAD.
— Mariana Barata, CEDOC - NOVA Medical School, is the co-author of this comment.
References:
Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, Harold D, Schrijvers EM, Ramirez-Lorca R, Debette S, Longstreth WT, Janssens AC, Pankratz VS, Dartigues JF, Hollingworth P, Aspelund T, Hernandez I, Beiser A, Kuller LH, Koudstaal PJ, Dickson DW, Tzourio C, Abraham R, Antunez C, Du Y, Rotter JI, Aulchenko YS, Harris TB, Petersen RC, Berr C, Owen MJ, Lopez-Arrieta J, Varadarajan BN, Becker JT, Rivadeneira F, Nalls MA, Graff-Radford NR, Campion D, Auerbach S, Rice K, Hofman A, Jonsson PV, Schmidt H, Lathrop M, Mosley TH, Au R, Psaty BM, Uitterlinden AG, Farrer LA, Lumley T, Ruiz A, Williams J, Amouyel P, Younkin SG, Wolf PA, Launer LJ, Lopez OL, van Duijn CM, Breteler MM, . Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010 May 12;303(18):1832-40. PubMed.
Tampellini D, Gouras GK. Synapses, synaptic activity and intraneuronal abeta in Alzheimer's disease. Front Aging Neurosci. 2010;2 PubMed.
Di Paolo G, Sankaranarayanan S, Wenk MR, Daniell L, Perucco E, Caldarone BJ, Flavell R, Picciotto MR, Ryan TA, Cremona O, De Camilli P. Decreased synaptic vesicle recycling efficiency and cognitive deficits in amphiphysin 1 knockout mice. Neuron. 2002 Feb 28;33(5):789-804. PubMed.
Ubelmann F, Burrinha T, Salavessa L, Gomes R, Ferreira C, Moreno N, Guimas Almeida C. Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep. 2017 Jan;18(1):102-122. Epub 2016 Nov 28 PubMed.
Schürmann B, Bermingham DP, Kopeikina KJ, Myczek K, Yoon S, Horan KE, Kelly CJ, Martin-de-Saavedra MD, Forrest MP, Fawcett-Patel JM, Smith KR, Gao R, Bach A, Burette AC, Rappoport JZ, Weinberg RJ, Martina M, Penzes P. A novel role for the late-onset Alzheimer's disease (LOAD)-associated protein Bin1 in regulating postsynaptic trafficking and glutamatergic signaling. Mol Psychiatry. 2019 Apr 9; PubMed.
Make a Comment
To make a comment you must login or register.