An Axon Self-Destruct Button Triggers Energy Woes
Quick Links
Just as losing a limb can spare a life, parting with a damaged axon by way of Wallerian degeneration can spare a neuron. A protein called SARM1 acts as the self-destruct button, and now researchers led by Jeffrey Milbrandt of Washington University Medical School in St. Louis believe they have figured out how. They report in the April 24 Science that SARM1 forms dimers that trigger the destruction of NAD+. Basic biochemistry dictates that this enzyme cofactor is essential for cell survival.
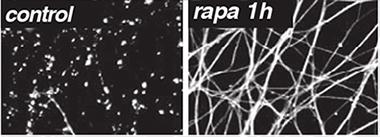
Severed axons normally degenerate within a day, but axons treated with rapamycin to destroy SARM1 persist. [Image courtesy of Science/AAAS]
SARM1 and NAD+ have emerged as key players in the complex, orderly process underlying Wallerian degeneration. Scientists are still filling in other parts of the pathway. SARM1, short for sterile alpha and TIR motif-containing 1, seems to act as a damage sensor, but researchers are not sure how. Recently, researchers led by Marc Tessier-Lavigne at Rockefeller University, New York, found that SARM1 turns on a mitogen-activated protein (MAP) kinase cascade that is involved (see Jan 2015 news; Osterloh et al., 2012). Loss of NAD+ may also contribute to axon degeneration, because its concentration drops in dying axons, and Wlds mutant mice that overproduce an NAD+ synthase have slower Wallerian degeneration (see Nov 2001 news; Wang et al., 2005).
Now, first author Josiah Gerdts confirms that SARM1 is the self-destruct switch. He engineered a version of the protein with a target sequence for tobacco etch virus (TEV) protease embedded in it. Using a rapamycin-activated form of TEV, he eliminated SARM1 from axons he had sliced off of mouse dorsal root ganglion (DRG) neurons. Without SARM1, the severed axons survived (see image above).
SARM1 contains SAM and TIR domains, which promote protein-protein interactions (Kang and Lee, 2011). Previously, Gerdts discovered that the TIR domain was sufficient to induce degeneration, even in healthy axons, but it relied on the SAM region to bring multiple SARM1 molecules together (Gerdts et al., 2013). He hypothesized that axonal SARM1 multimerizes upon axon damage. To test this idea, he used a standard biochemical technique to force the SARM1 TIR domains together. He fused domains to one or another of the rapamycin-binding peptides Frb and Fkbp and expressed them in DRG neurons (Fegan et al., 2010). When he added rapamycin to the cultures, the Frb and Fkbp snapped the TIR domains together within minutes. As Gerdts had predicted, this destroyed axons, confirming that SARM1 activates via dimerization.
Next, the authors investigated what happens to NAD+ during that process. Using high-performance liquid chromatography, Gerdts measured the concentration of NAD+ in the disembodied axons. Normally, its level dropped by about two-thirds within 15 minutes of severing. In axons from SARM1 knockout mice, however, the NAD+ concentration stayed unchanged. In neurons carrying the forced-dimerization constructs, adding rapamycin was sufficient to knock down NAD+ levels—Gerdts did not even have to cut the axons. Ramping up NAD+ production by overexpressing its synthases, NMNAT and NAMPT, overcame the effects of TIR dimerization, and the axons survived. Gerdts concluded that loss of NAD+ was a crucial, SARM1-controlled step on the way to degeneration.
He still wondered what caused the loss of NAD+. It might be that the axon simply stopped making it, or maybe the Wallerian pathway actively destroyed it. To distinguish between these possibilities, Gerdts added radiolabeled exogenous NAD+ to human embryonic kidney HEK293 cultures expressing the forced-dimerization TIR domains. Rapamycin caused them to rapidly degrade the radioactive NAD+, confirming that the cell actively disposes of it.
Gerdts suspects that with this essential cofactor gone, the axon runs out of energy and can no longer survive. He speculated that the MAP kinase cascade reportedly turned on by SARM1 might lead to NAD+ destruction. Alternatively, SARM1 might induce distinct MAP kinase and NAD+ destruction pathways in parallel, he suggested.
“Demonstrating how NAD+ is actively and locally degraded in the axon is a big advance,” commented Andrew Pieper of the Iowa Carver College of Medicine in Iowa City, who was not involved in the study. Jonathan Gilley and Michael Coleman of the Babraham Institute in Cambridge, U.K., predict that there will be more to the story. They note that a drug called FK866, which prevents NAD+ production, protects axons in some instances (Sasaki et al., 2009; Di Stefano et al., 2015). Gerdts suggested that FK866 acts on processes upstream of SARM1, delaying the start of axon degeneration. In contrast, his paper only addressed what happens after SARM1 activates. “It will be fascinating to see how the apparent contradictions raised by this new study will be resolved,” wrote Gilley and Coleman.
Could these findings help researchers looking for ways to prevent neurodegeneration? “The study supports the notion that augmenting NAD+ levels is potentially a valuable approach,” said Pieper. He and his colleagues developed a small molecule that enhances NAD+ synthesis, now under commercial development (see Sep 2014 news). It improved symptoms in ALS model mice, and protected neurons in mice mimicking Parkinson’s (see Oct 2012 news). NAD+ also activates sirtuin, an enzyme important for longevity and stress resistance as well as learning and memory (see Jul 2010 news; Sep 2007 news).
However, both Pieper and Gerdts cautioned that they cannot clearly predict which conditions might benefit from an anti-SARM1 or NAD+-boosting therapy. At this point, Gerdts said, researchers do not fully understand how much axon degeneration contributes to symptoms of diseases like Alzheimer’s and Parkinson’s. He suggested that crossing SARM1 knockout mice with models for various neurodegenerative conditions would indicate how well an anti-Wallerian therapy might work.—Amber Dance
References
News Citations
- MAPping Death Pathways in Axons
- Protein Chimera Found to Protect Axons from Degeneration
- Neuron-Protecting P7C3 Compounds Take Steps Toward the Clinic
- Compound to the Rescue in Parkinson’s, ALS Models
- Research Brief: SIRTs Keep Brain Minty Fresh
- Cells Find Inner Strength in Mitochondrial NAD+
Paper Citations
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, Macdonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH, Conforti L, Coleman M, Tessier-Lavigne M, Züchner S, Freeman MR. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012 Jul 27;337(6093):481-4. PubMed.
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005 Aug 1;170(3):349-55. PubMed.
- Kang JY, Lee JO. Structural biology of the Toll-like receptor family. Annu Rev Biochem. 2011;80:917-41. PubMed.
- Gerdts J, Summers DW, Sasaki Y, DiAntonio A, Milbrandt J. Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J Neurosci. 2013 Aug 14;33(33):13569-80. PubMed.
- Fegan A, White B, Carlson JC, Wagner CR. Chemically controlled protein assembly: techniques and applications. Chem Rev. 2010 Jun 9;110(6):3315-36. PubMed.
- Sasaki Y, Vohra BP, Lund FE, Milbrandt J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci. 2009 Apr 29;29(17):5525-35. PubMed.
- Di Stefano M, Nascimento-Ferreira I, Orsomando G, Mori V, Gilley J, Brown R, Janeckova L, Vargas ME, Worrell LA, Loreto A, Tickle J, Patrick J, Webster JR, Marangoni M, Carpi FM, Pucciarelli S, Rossi F, Meng W, Sagasti A, Ribchester RR, Magni G, Coleman MP, Conforti L. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 2015 Apr;22(5):731-42. Epub 2014 Oct 17 PubMed.
Further Reading
Papers
- Feng Y, Yan T, He Z, Zhai Q. Wld(S), Nmnats and axon degeneration--progress in the past two decades. Protein Cell. 2010 Mar;1(3):237-45. PubMed.
- Yan T, Feng Y, Zhai Q. Axon degeneration: Mechanisms and implications of a distinct program from cell death. Neurochem Int. 2010 Mar;56(4):529-34. PubMed.
- Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, Magni G, Glass JD, Coleman MP. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ. 2007 Jan;14(1):116-27. PubMed.
Primary Papers
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. Neurobiology. SARM1 activation triggers axon degeneration locally via NAD⁺ destruction. Science. 2015 Apr 24;348(6233):453-7. Epub 2015 Apr 23 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Babraham Institute
Babraham Institute
Progress in science usually comes from considering two apparently contradictory findings and asking how both can be correct. On this occasion there are two such results. One is the striking data reported by Gerdts and colleagues indicating that artificial dimerization of the SARM1 TIR domain (sTIR) can deplete NAD and induce axon degeneration and cell death. The other data, which were already published by at least three groups, indicated that a drug that depletes NAD, FK866, phenocopies WLDS expression: It preserves axons in several contexts, including injured neurites in primary culture, explanted nerve-muscle preparations, and severed axons in zebrafish embryos (Sasaki et al., 2009; Di Stefano et al., 2014; Shen et al., 2014). Gerdts et al. cite one of these papers from their own group (Sasaki et al., 2009) in a manner unrelated to neuroprotection, while not citing the others. Consequently, what we consider to be the most interesting implications of these data are not discussed.
It is puzzling how SARM1-mediated depletion of NAD relates to the rapid loss of nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) and to alterations in NAD and other related metabolites in injured axons/nerves. NMNAT2 appears to be the major NMNAT isoform in axons and is essential for their survival (Gilley and Coleman, 2010). Because NMNAT2 has a short half-life, and since NAD is constantly turned over, loss of the enzyme in injured axons could itself be sufficient to underlie the decline in NAD. Crucially, a rise in the NMNAT substrate, NMN, which seems to be pro-degenerative, is also seen in injured nerves while the NMNAT product, NAD, falls (Di Stefano et al., 2014). This rise in NMN fits less easily with SARM1-mediated NAD depletion being the sole driver of axon degeneration. NMN would only accumulate if the enzyme needed to take it away (NMNAT, and probably specifically NMNAT2) was missing. In fact, NAD levels decline substantially in the absence of SARM1 within five days of nerve injury, though the axons continue to survive for over two weeks (Gilley et al., 2015). Interestingly, mice lacking NMNAT2 die at birth due to an extensive axon defect (Gilley et al., 2013), and we have recently shown that SARM1 depletion totally rescues this phenotype, allowing mice to survive and remain healthy up to at least 12 months of age. However, SARM1 depletion seems to work in these mice via a mechanism that appears largely independent of changes to NAD (or NMN) levels (Gilley et al., 2015). Hence, Gerdts et al.'s finding that NAD levels are partially maintained in the absence of SARM1 30h after a nerve lesion does not easily explain the survival of NMNAT2 null mice for up to 12 months when SARM1 is also removed. This, too, suggests there is more to the story.
What the field now needs is a balanced review (or several reviews from different authors) on this topic that attempts to reconcile these different findings. The history of this field shows that this is how progress is made. The 2004 publication of axon preservation by heavily overexpressed NMNAT1 in primary culture (Araki et al., 2004), followed by findings that NMNAT1 does not preserve axons in transgenic mice (Conforti et al., 2007; Yahata et al., 2009), were key factors leading to the identification of NMNAT2 as an endogenous axon survival factor (Gilley and Coleman, 2010). This led us to propose an attractive and simple model in which the aberrant WLDS fusion protein, or mislocalized NMNAT1 (due to high overexpression or mutation), deliver stable NMNAT1 activity into axons (where it is not normally found) to functionally substitute for loss of labile NMNAT2. It will be fascinating to see how the apparent contradictions raised by this new study resolve themselves this time.
Alzheimer’s disease involves massive loss of synapses and white matter and readers of this forum will be aware that it is important in the longer term to know whether the Wallerian axon degeneration pathway is at least partially responsible for this. This is a more complex question, but progress toward understanding the NMNAT/SARM1 pathway is an essential first step. Eventually the aim will be to ask whether markers and modifiers of this pathway play key roles in animal models, and ultimately human cases, of Alzheimer’s disease.
References:
Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004 Aug 13;305(5686):1010-3. PubMed.
Conforti L, Fang G, Beirowski B, Wang MS, Sorci L, Asress S, Adalbert R, Silva A, Bridge K, Huang XP, Magni G, Glass JD, Coleman MP. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ. 2007 Jan;14(1):116-27. PubMed.
Di Stefano M, Nascimento-Ferreira I, Orsomando G, Mori V, Gilley J, Brown R, Janeckova L, Vargas ME, Worrell LA, Loreto A, Tickle J, Patrick J, Webster JR, Marangoni M, Carpi FM, Pucciarelli S, Rossi F, Meng W, Sagasti A, Ribchester RR, Magni G, Coleman MP, Conforti L. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 2015 Apr;22(5):731-42. Epub 2014 Oct 17 PubMed.
Gilley J, Adalbert R, Yu G, Coleman MP. Rescue of peripheral and CNS axon defects in mice lacking NMNAT2. J Neurosci. 2013 Aug 14;33(33):13410-24. PubMed.
Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010 Jan 26;8(1):e1000300. PubMed.
Gilley J, Orsomando G, Nascimento-Ferreira I, Coleman MP. Absence of SARM1 Rescues Development and Survival of NMNAT2-Deficient Axons. Cell Rep. 2015 Mar 31;10(12):1974-81. Epub 2015 Mar 26 PubMed.
Sasaki Y, Vohra BP, Lund FE, Milbrandt J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci. 2009 Apr 29;29(17):5525-35. PubMed.
Sasaki Y, Vohra BP, Baloh RH, Milbrandt J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J Neurosci. 2009 May 20;29(20):6526-34. PubMed.
Shen H, Hyrc KL, Goldberg MP. Maintaining energy homeostasis is an essential component of Wld(S)-mediated axon protection. Neurobiol Dis. 2013 Nov;59:69-79. Epub 2013 Jul 24 PubMed.
Yahata N, Yuasa S, Araki T. Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J Neurosci. 2009 May 13;29(19):6276-84. PubMed.
View all comments by Jonathan GilleyMake a Comment
To make a comment you must login or register.