BACE1 Conditional Knockouts Model Adult BACE Inhibition
Quick Links
A new conditional knockout mouse offers clues on the effects of dialing down β-secretase, a.k.a. BACE1, a target for potential Alzheimer’s therapies. Researchers led by Riqiang Yan, Lerner Research Institute, Cleveland, report in the February 14 Journal of Experimental Medicine that the mice, which gradually lose expression of BACE1 soon after birth, appear healthy but for weakened neural plasticity. When crossed to a mouse model of AD, their postnatal reduction in BACE1 not only halted Aβ plaque formation, but also appeared to eliminate existing plaques. This led to claims in the press that AD can be completely reversed by targeting BACE (e.g., Feb 15 Newsweek), which came two days after Merck announced the termination of its BACE inhibitor verubecestat (Feb 2018 news). Alzforum covered preliminary data from Yan’s study in 2016, when he presented them at the second Kloster Seeon meeting on BACE proteases in health and disease in Germany (Oct 2016 news).
- In a conditional BACE knockout, expression begins falling in early adulthood.
- Apart from weak LTP, knockouts appear healthy.
- When crossed with 5xFAD mice, the knockout appears to ablate amyloid plaques.
“I am delighted to see that someone finally made the conditional BACE1 knockout mouse,” said Joanna Jankowsky, Baylor College of Medicine in Houston. Stefan Lichtenthaler, German Center for Neurodegenerative Diseases in Munich, was also enthusiastic. “It is particularly intriguing to see that BACE1 inhibition not only reduces plaque growth, but may even shrink existing plaques,” he wrote (see full comment below). However, researchers noted important limitations of this study, as well.
BACE1 embryonic knockouts develop brain and muscle defects, display abnormal behaviors, and often die young (Vassar et al., 2014; Barão et al., 2016; Hou et al., 2017). Researchers developing therapies that target BACE1 have long wondered to what extent these problems stem from developmental errors versus harmful effects later in life.
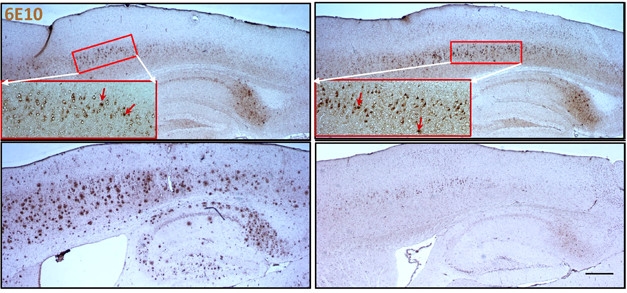
Vanishing Act? From postnatal day 75 (top) to 300 (bottom), brain amyloid plaques (red arrows) grow in number and size in 5xFAD mice (left)), but dwindle in 5xFAD mice crossed to BACE1 conditional knockouts (right). [© Hu et al., J Exp Med, 2018.]
To address this question, first author Xiangyou Hu engineered an inducible BACE1 knockout. He used a tamoxifen-inducible cre driven by the ubiquitin promoter, which is active in nearly all cells, to control the removal of exon 2 from the BACE gene. To the researchers’ surprise, even before giving these animals tamoxifen, brain BACE1 levels started dropping around 30 days after birth, falling to 50 percent at two months, and to 20 percent at four months of age. “The cre expression turned out to be very leaky,” explained Yan, now at the University of Connecticut, Farmington. Though eliminating the model’s manipulability, this technical fluke had a silver lining in that it avoided potential side effects from tamoxifen.
Joining a collection of BACE transgenic mouse models, these new conditional knockouts (cKOs) have a normal lifespan and, so far, appear free of the deficits that afflict embryonic KOs. Five-month-old cKOs sport well-myelinated axons in their corpora callosa, sciatic nerves, and optic nerves, and the astrocytes in their dentate gyri are similar in number and type to those of healthy controls; all these features are known to be disrupted in embryonic knockouts. Also, unlike embryonic KOs, the cKOs do not experience spontaneous seizures. However, the cKOs have elevated levels of full-length neuregulin, a BACE1 substrate involved in brain development and synaptic plasticity. Perhaps because of this, long-term potentiation evoked by stimulation in the hippocampus was lower than LTP by about half in wild-type controls.
To model the consequences of losing BACE1 in AD, the researchers crossed the cKOs with 5xFAD mice, which start accumulating amyloid plaques at 2.5 months of age. They compared plaque pathology using the 6E10 antibody, which recognizes plaques, APP, and soluble Aβ peptides. At 2.5 months, plaques in the cortices of the 5xFAD/BACE cKO mice numbered, on average, 24 per sagittal slice, compared with 5xFAD controls. Plaques steadily grew in size and number in the controls. By 10 months of age, plaques numbered almost 800 in the controls, but were essentially undetectable if BACE was absent (see image above).
“The brain has some system to eliminate amyloid plaques if you turn off the spigot,” said Yan. This idea has been tested before, with mixed results. Suppressing γ-secretase reduced pre-existing amyloid plaques in APP transgenics, whereas suppressing APP itself did not (Jul 2005 news; Jankowsky et al., 2005; Mar 2013 news).
Jochen Herms, German Center for Neurodegenerative Diseases, Munich, recently reported that the BACE1 inhibitor NB-360 blocks new plaque formation but can’t eliminate existing plaques (Jan 2018 news). Herms was not convinced plaques had vanished in Yan’s new crosses. He believes the fading of the 6E10 signal might reflect reduced intraneuronal accumulation of APP fragments, since 6E10 binds not only Aβ, but also BACE cleavage products such as C99. “The [loss of 6E10 binding] does not necessarily mean that existing plaques have been cleared,” Herms said, adding that it is difficult to understand exactly what is happening since, for example, one would expect intracellular, full-length APP to increase with BACE loss. Both he and Jankowsky would have liked to see a more specific marker for fibrillary Aβ aggregates used. Yan told Alzforum he obtained essentially identical findings using thioflavin S, which binds to fibrillary plaques. However, he analyzed only a few such samples, and these data were not included in the paper.
As plaques dwindled in the cKO/5xFAD mice, gliosis as per anti-GFAP antibodies and dystrophic neurites detected by anti-RTN3 antibodies both faded. Moreover, the weak hippocampal LTP seen in 5xFAD mice was partially rescued. After stimulation, excitatory postsynaptic potentials climbed by 21 percent in the crosses, compared with 5 percent in the 5xFAD mice. That’s despite weakened LTP seen in the BACE cKO alone. By comparison, LTP increases 60 percent in wild-type mice, and 28 percent in BACE cKOs.
What do the findings mean for the clinical future of BACE1 inhibitors? Ulf Neumann, Novartis, Basel, Switzerland, noted that the cKO/5xFAD mice were still young when BACE1 dipped more than 50 percent. “They may produce Aβ deposits that are ‘easy’ to clear, with comparatively few densely compact plaques,” he said. In contrast, AD patients who have harbored plaques for many decades may have irreversible damage, he said. While he thinks there are caveats in interpreting the data, Neumann said the study can be taken as another hint that early, and very early, anti-Aβ treatments have chances of success.
Yan acknowledged that the mice’s young age is a limitation. “We are now making additional cre lines to control BACE1 expression more tightly in late adult stages,” he said.—Marina Chicurel
References
Therapeutics Citations
News Citations
- Merck Axes Verubecestat for Prodromal AD, Researchers Say ‘Go Earlier’
- What Exactly Does BACE Do in Adults?
- Amyloid Hypothesis—Closing the Spigot Helps, Temporarily
- Inducible APP Mice—Cognition Restored in Advanced Amyloidosis?
- BACE Block Nips New Plaques in the Bud, Old Ones Keep Growing
Research Models Citations
Antibody Citations
Paper Citations
- Vassar R, Kuhn PH, Haass C, Kennedy ME, Rajendran L, Wong PC, Lichtenthaler SF. Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J Neurochem. 2014 Jul;130(1):4-28. Epub 2014 Apr 19 PubMed.
- Barão S, Moechars D, Lichtenthaler SF, De Strooper B. BACE1 Physiological Functions May Limit Its Use as Therapeutic Target for Alzheimer's Disease. Trends Neurosci. 2016 Mar;39(3):158-69. Epub 2016 Jan 30 PubMed.
- Hou H, Fan Q, He W, Suh H, Hu X, Yan R. BACE1 Deficiency Causes Abnormal Neuronal Clustering in the Dentate Gyrus. Stem Cell Reports. 2017 Jul 11;9(1):217-230. Epub 2017 Jun 29 PubMed.
- Jankowsky JL, Slunt HH, Gonzales V, Savonenko AV, Wen JC, Jenkins NA, Copeland NG, Younkin LH, Lester HA, Younkin SG, Borchelt DR. Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease. PLoS Med. 2005 Dec;2(12):e355. Epub 2005 Nov 15 PubMed.
Other Citations
External Citations
Further Reading
No Available Further Reading
Primary Papers
- Hu X, Das B, Hou H, He W, Yan R. BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J Exp Med. 2018 Mar 5;215(3):927-940. Epub 2018 Feb 14 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
German Center for Neurodegenerative Diseases (DZNE)
This is a very interesting study that provides several important advances and insights for the field. It is particularly intriguing to see that BACE1 inhibition not only reduces plaque growth, but may even shrink existing plaques. If it also happens in patients this would be fantastic news for the clinical trials with BACE inhibitors. Because AD mouse models, including the 5xFAD model used in this study, typically mimic presymptomatic AD, it would appear possible that secondary prevention trials with BACE inhibitors may not only yield reduced growth or number of plaques, but even a shrinking of pre-existing plaques and a reduction of neuroinflammation—provided that the drugs are given early enough before the symptoms start. This should and will be tested in the trials’ participants using PET imaging. These new findings contradict a recent study that demonstrated that plaques remain stable in size when BACE1 is inhibited pharmacologically. I am sure that this discrepancy will be resolved with future experiments and may depend on the mouse line used, or the level or duration of BACE1 inhibition.
We also need to consider that the amazing effects of the mouse study were accompanied by an intriguingly gradual reduction of BACE1 protein levels. It is difficult to predict exactly which inhibition level of BACE1 would be needed in humans to achieve the same results. Potentially, the levels currently used in trials are already sufficient.
Another take-home message of the study is that memory and LTP deficits were improved in the AD mice as BACE1 was suppressed. While this is good news, the study also demonstrated that LTP did not fully recover. This indicates that the therapeutically desired BACE inhibition in adult mice may interfere with physiological BACE1 functions. Besides LTP, this includes muscle spindle formation/maintenance and dendritic spine densities in adult mice. Translated to humans, this could indicate a larger number of falls or psychiatric symptoms in individuals treated with BACE inhibitors. Whether this is a realistic concern will be seen from the results of the currently ongoing and recently terminated BACE inhibitor trials.
Constitutive BACE1-deficient mice show a number of different phenotypes. Another major insight of the new study is that adult deletion of BACE1 overcomes at least one of the symptoms—the hypomyelination. The new conditional BACE1-deficient mice are an excellent tool to analyze whether the other BACE1-deficient mouse phenotypes are also of developmental origin and which functions of BACE1 are relevant in adult mice. Taken together, the new study is an excellent basis both for basic and translational BACE1 research.
I am interested to know why, under BACE1 cKO, there is, in addition to an expected decrease in C99, a decrease in C83 as well. The authors suggest that a cKO of BACE1 improves autophagy, increasing turnover of C-terminal fragments. However, there is no change in C83 in the original KO (Cai, 2001). Have the authors looked at APP trafficking in their model? I would be interested to know if the amount of sAPPα is the same, i.e. is APP still being trafficked to the cell surface?
References:
Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001 Mar;4(3):233-4. PubMed.
University of Connecticut Health
The collaborative team of Drs. Herms and Neumann published a nice study on the use of NB-360 to block formation of new plaques. The main difference between their study and ours is the chemical inhibition of versus the sequential deletion of BACE1 in AD mouse models. It is understood that chemical inhibition of BACE1 may never reach the level of near-complete deletion of BACE1 in mouse brains. In mice with conditional deletion of BACE1, BACE1 protein was barely detectable and its activity is diminished to the extent of drastically decreased APP-C99 and Aβ levels.
The reason for the reversal of amyloid plaques, evidenced not only by 6E10 staining of plaques but also staining by thioflavin S and an anti-Aβ42 antibody (AB-2341375 from IBL), is not fully understood yet, but is actively being investigated in the lab. As pointed out by Marc Tambini, it is intriguing and counterintuitive to see the observed reduction of APP-C83. Presenting a counterintuitive result is important for motivating a novel finding. When we first overexpressed BACE1 in cultured cells expressing Swedish APP, we actually observed reduction of Aβ1-40 and Aβ1-42 levels rather than an expected increase (Figure 4a in Yan et al. 1999), and this counterintuitive result led to the discovery of β'-cleavage.
References:
Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999 Dec 2;402(6761):533-7. PubMed.
Make a Comment
To make a comment you must login or register.