C1q Shows Promise as Therapeutic Target to Stop Synapse Loss
Quick Links
An old insight in neurodegenerative research is that while a given disease is visibly characterized by protein deposits in the brain, synapse loss is what correlates most closely with the disease's signature functional decline, be it cognitive or motor. A new paradigm for tackling this dichotomy is to try to treat these diseases by blocking complement protein C1q, which tags synapses for elimination by glia.
- A C1q antibody hit its target in a Phase 2 Huntington’s trial.
- A small molecule that indirectly blocks C1q restored synaptic density and memory in amyloidosis mice.
- The data strengthen the case for targeting C1q in neurodegenerative disease.
Recent clinical and preclinical data strengthen the case for this approach. On the clinical side, biotech startup Annexon, based in Brisbane, California, recently reported positive Phase 2 results for an anti-C1q antibody. In a small, open-label trial of Huntington’s patients, the drug suppressed C1q in cerebrospinal fluid, suggesting good target engagement. Tantalizingly, participants’ clinical status remained stable over six months of dosing and three months of follow-up, though without a placebo control, researchers can draw no conclusions about efficacy.
Tiago Mestre at the University of Ottawa, Ontario, Canada, urged caution in interpreting the trial results, but said they support further studies. “Overall, it is exciting news that a new compound warrants further investigation to address the enormous gap of disease modification in HD,” he wrote (full comment below).
Meanwhile, researchers led by Stephen Strittmatter and Zhengxin Cai at Yale University in New Haven, Connecticut, took a different angle, keeping C1q out of synapses in amyloidosis mice by using a small molecule that blocks aberrant Aβ-oligomer signaling through the metabotropic glutamate receptor 5 (mGluR5). In the June 1 Science Translational Medicine, they reported that synaptic density rebounded after treatment, and learning and memory returned to normal. This compound is in Phase 1 testing.
“I was very impressed by this paper,” Andrea Tenner at the University of California, Irvine, told Alzforum. “The study is well-designed and really exciting. This [compound] is a potential candidate for the treatment toolbox.” Gek-Ming Sia at the University of Texas, San Antonio, agreed. “[The paper] provides further evidence that C1q is a promising therapeutic target in neurodegenerative diseases,” he wrote to Alzforum (comment below).
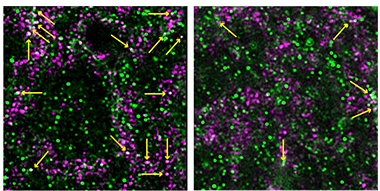
C1q Begone. In the hippocampi of APP knock-in mice (left), C1q (pink) loiters in post-synapses (green; overlay looks yellow, arrows). Treatment with an mGluR5 modulator (right) chases C1q away. [Courtesy of Spurrier et al., Science Translational Medicine/AAA.]
C1q’s role in synapse elimination was first identified by the late Ben Barres of Stanford University, Palo Alto, California, and Beth Stevens, now at Boston Children’s Hospital (Nov 2007 conference news; Dec 2007 news). They found the brain uses C1q to prune weak synapses during development, but this process aberrantly reactivates in neurodegenerative disease (Mar 2015 conference news; Nov 2015 conference news; Apr 2016 news). Later work on amyloidosis and HD model mice showed that suppressing or blocking key complement proteins preserved synapses and improved memory or motor skills, respectively (Jun 2017 news; Jul 2019 conference news).
In 2011, Barres co-founded Annexon to explore complement-based therapies. The C1q antibody, dubbed ANX005, is being tested for HD and amyotrophic lateral sclerosis, as well as some autoimmune disorders (Lansita et al., 2017). The trial enrolled 28 people with early HD, all of whom received ANX005 by intravenous infusion every two weeks for six months. Treatment squelched C1q in the serum and CSF to below detectable levels. In a press briefing, Ted Yednock at Annexon noted that downstream complement proteins such as C3 and C4a stayed low even after treatment stopped, suggesting the treatment suppressed inflammation. “We think we can look at lower and less frequent dosing,” Yednock said.
The trial was not powered to determine efficacy. Nonetheless, the scientists noted that participants seemed to maintain their cognitive and motor abilities better than would be expected from historical disease-progression data. In particular, when researchers split the cohort in half based on whether they had greater or less than the median CSF C4a level at baseline, they spotted a separation. C4a is a marker of activated complement. The group with more C4a improved on functional endpoints on average, while the group with less C4a declined. Numerically, 75 percent of people in the high C4a group improved with treatment, compared with 36 percent of the low C4a group. CEO Douglas Love noted that Annexon is discussing with regulators whether to recruit only people with high baseline complement for future trials.
The trial brought up some safety signals. Three participants dropped out due to adverse events—lupus, pneumonia, and anemia—that were judged to be related to treatment. At baseline, these three participants had high levels of antinuclear antibodies, i.e., autoantibodies that target nuclear proteins. Love noted that future trials will exclude people with high ANA titers, who amount to about 20 percent of the population.
Mestre said the number of treatment-emergent serious adverse events was high for a trial of this size. He also suggested researchers reserve judgment on clinical effects. “We know from HD and other neurodegenerative disorders how the power of placebo can be reflected in efficacy outcomes, even after months of treatment,” Mestre wrote.
For their part, Strittmatter and colleagues were not looking for a way to target C1q. They happened upon it in the course of investigating the role of glutamate signaling through mGluR5 in Alzheimer’s disease. To do this, they made use of BMS-984923, a compound Bristol Myers Squibb developed as part of a schizophrenia research program that has since been discontinued. BMS was trying to generate activators of mGluR5, but BMS-984923 turned out to be a silent allosteric modulator (SAM); that is, it did not bind mGluR5’s active site and had no effect on glutamate signaling.
When Strittmatter and colleagues tested it, they discovered an unexpected property: BMS-984923 prevented mGluR5 from binding cellular prion protein complexed with oligomeric Aβ. Notably, feeding the compound to APP/PS1 mice for a month preserved normal synaptic density and memory, hinting at potential to treat AD (Haas et al., 2017).
In the new study, the researchers dug into the SAM's mechanism of action. Joint first authors Joshua Spurrier, LaShae Nicholson, Xiaotian Fang, and Austin Stoner performed single-nuclei RNA-Seq on cortical and hippocampal samples from treated and untreated wild-type, APP/PS1, and APPNL-G-F knock-in mice. SAM treatment normalized about 70 percent of the dysregulated neuronal genes in the AD models, but had little effect on glial genes. Many of the corrected genes were synaptic, suggesting a broad rescue of synaptic function.
How did synapses change? Lo and behold, immunostaining revealed that post-synaptic C1q, which climbs about 50 percent in the AD mice, dropped back to wild-type levels with treatment (see image above). This occurred even though the overall amount of C1q in treated AD mice stayed unchanged, at about fivefold higher than in wild-types.
In addition, astrocytes contained about 20 percent fewer synaptic proteins after SAM treatment, suggesting they had engulfed fewer synapses. The authors focused on astrocytes rather than microglia because of a previous paper suggesting the former specialize in synapse cleanup, whereas the latter dispose of the neuron's soma (Jun 2020 news). Notably, SAM treatment did not affect gliosis or plaque load.
Strittmatter believes the findings support a model wherein blocking aberrant signaling by Aβ oligomers restores normal neuronal function, resulting in the neurons expressing fewer “eat me” or more “don’t eat me” signals at their synapses. That, in turn, prevents C1q from binding.
What those signals might be is not clear. One possibility is the “don’t eat me” signal CD47, which was rescued by SAM treatment (Dec 2021 news). Previous research has identified synaptic p-tau as an “eat me” signal; intriguingly, SAM treatment dropped levels of p-tau205 and p-tau217, although they remained higher than in wild-type mice (Jul 2018 conference news; Aug 2019 news).
Importantly, treatment did more than halt synapse loss. One month of SAM treatment restored synaptic density to wild-type levels, as seen by before-and-after synaptic PET imaging, as well as by directly measuring co-localization of pre- and post-synaptic proteins by SEQUIN (Jun 2020 news). This synaptic recovery persisted for at least a month after the drug was washed out. The findings support the idea that new synapse formation continues even during amyloidosis, and offer hope that cognitive recovery could be possible in the early stages of disease, before neurons die, Strittmatter noted.
Martin Kerschensteiner at Ludwig-Maximilians University, Munich, said that measuring synaptic density in the same mice before and after treatment was methodologically elegant, helping to control for the individual variability in synapse loss with disease. “It gives a convincing demonstration of synapse protection,” he told Alzforum.
Other data showed that BMS-984923 has good drug properties. It is a small lipophilic molecule that enters the brain readily, achieving twice the concentration in mouse brain than in plasma. Because it is a silent allosteric modulator, it does not interfere with physiological glutamate signaling through mGluR5. In mice, it had no detectable toxicity at 250 times the effective dose, suggesting it could be well-tolerated.
Yale researchers are testing BMS-984923 in a Phase 1 single-ascending-dose study in 36 healthy older adults. Results from this publicly funded trial are expected soon. Meanwhile, BMS has licensed the drug to Yale biotech startup Allyx Therapeutics, which will run any future trials, in Alzheimer's disease.
Other researchers were enthusiastic about the approach, noting some advantages over hitting C1q on the nose with an antibody. Tenner said C1q can be neuroprotective early in neurodegeneration, making direct inhibition problematic. She also cautioned that knocking out C1q systemically could have negative effects such as autoimmunity.
Kerschensteiner found the SAM's broad effects encouraging. “Not only does it protect synapses, but it also interferes with some aspects of Aβ-mediated pathology such as p-tau,” he said. “If you just blocked C1q, you would not affect pathology inside the neuron.”—Madolyn Bowman Rogers
References
News Citations
- San Diego: MHC Class I and Complement—Holding Down Second Jobs in the Synapse
- Paper Alert: Does the Complement Devour Synapses?
- Microglia Rely on Mixed Messages to Select Synapses for Destruction
- Microglia Control Synapse Number in Multiple Disease States
- Paper Alert: Microglia Mediate Synaptic Loss in Early Alzheimer’s Disease
- Sans Complement: Amyloid Grows, Synapses and Memory Stay
- Do Microglia Finish Off Stressed Neurons Before Their Time?
- Too Phatal: How Microglia, Astrocytes Snuff Out Dying Neurons
- Single Synapse Mass Spec Snags CD47 as Alzheimer’s Resilience Factor
- Synaptic Tau Clangs the Dinner Bell for Hungry Microglia
- Nixing Complement Protein Protects Neurons in Tauopathy Model
- Shiny SEQUIN: New Technique Counts Synapses Over Large Brain Volumes
Research Models Citations
Paper Citations
- Lansita JA, Mease KM, Qiu H, Yednock T, Sankaranarayanan S, Kramer S. Nonclinical Development of ANX005: A Humanized Anti-C1q Antibody for Treatment of Autoimmune and Neurodegenerative Diseases. Int J Toxicol. 2017 Nov/Dec;36(6):449-462. Epub 2017 Dec 4 PubMed.
- Haas LT, Salazar SV, Smith LM, Zhao HR, Cox TO, Herber CS, Degnan AP, Balakrishnan A, Macor JE, Albright CF, Strittmatter SM. Silent Allosteric Modulation of mGluR5 Maintains Glutamate Signaling while Rescuing Alzheimer's Mouse Phenotypes. Cell Rep. 2017 Jul 5;20(1):76-88. PubMed.
External Citations
Further Reading
Primary Papers
- Spurrier J, Nicholson L, Fang XT, Stoner AJ, Toyonaga T, Holden D, Siegert TR, Laird W, Allnutt MA, Chiasseu M, Brody AH, Takahashi H, Nies SH, Cañamás AP, Sadasivam P, Lee S, Li S, Zhang L, Huang YH, Carson RE, Cai Z, Strittmatter SM. Reversal of synapse loss in Alzheimer mouse models by targeting mGluR5 to prevent synaptic tagging by C1Q. Sci Transl Med. 2022 Jun;14(647):eabi8593. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Ottawa
The most interpretable result of the Phase 2 trial of the experimental complement inhibitor compound ANX005 in patients with Huntington’s disease (HD) is that there is target engagement for the proposed mechanism of C1q Target Inhibition.
Safety data warrants some reflection for future trials considering the total number of discontinuations and those explained by treatment-emergent serious adverse events in such a small trial.
The efficacy results should be interpreted with abundant reservation, as the trial was not designed to determine efficacy. This is a small, open-label trial, with no placebo arm in subjects at risk of or with early stage Huntington’s disease. We know from HD and other neurodegenerative disorders how the power of placebo can be reflected in efficacy outcomes, even after months of treatment.
In addition, any extrapolation of clinical and biomarker data on disease progression data from other HD cohorts needs to take into consideration that study participants were at a very early stage of the disease.
Overall, it is exciting news that a new compound warrants further investigation to address the enormous gap of disease modification in HD, and the HD community will follow the next chapter in its history with enthusiasm. Likely the company is advancing for a larger Phase 3 trial to address the question of efficacy of ANX005 that the current trial data is not able to solve.
University of Texas Health at San Antonio
The Spurrier study shows that an mGluR5-targeted AD therapeutic can normalize neuronal gene signatures and reduce C1q synaptic localization without altering total C1q amount, which further reinforces the concept that neuronal genes can regulate complement and microglia-mediated synapse loss.
This important work also highlights the gap in our understanding of how C1q is recruited to the synapse, and provides further evidence that C1q is a promising therapeutic target in neurodegenerative diseases.
Children's Hospital
Boston Children's Hospital
In their study, Spurrier et al., follow up on earlier work carried out by the same group (Haas et al., 2017). They recapitulate previous observations that treatment with a silent allosteric modulator of mGluR5 can increase synaptic density in AD mouse models, as well as rescue a variety of behavioral phenotypes. They extend this finding to other relevant disease models and expand their previous search into the underlying cellular and molecular mechanisms driving these beneficial phenotypes.
The strength of the current study lies in the pharmacokinetic profiling of BMS-984923, including investigations of receptor occupancy in nonhuman primates and studies of selectivity and toxicity across multiple species.
Furthermore, from a translational perspective, the demonstration in a preclinical rodent model that a key pathological hallmark of AD, namely the loss of synaptic populations, can to some extent be rescued by treatment with BMS-984923 and that this can be observed non-invasively with a clinically relevant SV2A PET tracer is another important finding. Imaging with this tracer in patients with neurodegenerative or neurodevelopmental disorders has already shown statistically significant associations between SV2A signal and clinically relevant disease transitions (Delva et al., 2022; Onwordi et al., 2020).
The authors' efforts to uncover mechanisms that might underlie the beneficial actions of BMS-984923 have yielded some interesting questions. At the same time, the data they present showing that C1Q association with synaptic proteins is diminished in AD models following treatment with BMS-984923—while itself intriguing, particularly in light of other work showing this protein to be important in synaptic elimination in other contexts—is insufficient on its own to support the claim that the reversal of synapse loss is mediated through this mechanism. The authors themselves acknowledge this in their discussion.
The authors have shown that synapse loss can be reversed and C1Q association with synaptic proteins reduced following treatment with BMS-984923, but these observations are only correlative. One way to test the necessity of reducing C1Q deposition with respect to the restoration of synaptic numbers would be to block this process through conditional genetic ablation of C1Q. Another would be to use pharmacological approaches, either together with, or in the absence of, the SAM, and to then assess whether blocking complement deposition alone is sufficient to rescue synapse density, as has been demonstrated in more acute paradigms earlier in disease progression and following genetic ablation of complement components from birth, and to assess whether there are any additive or synergistic effects of treatment with both agents.
It would also be interesting to look at how BMS-984923 might be acting to inhibit C1Q association with synaptic proteins or other functions of C1Q independent of classical complement pathway activation. Are downstream signaling or activation events in the complement cascade reversed or impacted by this compound?
Some interesting questions that arise not only from this work but also that of others are: What molecular mechanisms drive enhanced C1Q association with synaptic proteins in AD models? How specific is this phenotype to certain types of synapses or synaptic populations, and how are these associations reversed or impacted by treatment with BMS-984923? Given the role neuronal activity has been shown to play in microglial engulfment during developmental pruning events, could it be that altered patterns of activity within circuits are in some way acting as an instructive cue to prompt this process? This is intriguing given these authors' previous work showing the normalization of oligomeric Aβ-induced impaired LTD phenotypes in slice culture following treatment with BMS-984923 (Haas et al., 2017).
Finally, a caveat of the conclusions the authors draw is that mGluR5 is also expressed by specific populations of astrocytes under certain contexts. This includes AD models, where it has been implicated in synaptogenesis and changes in neuronal activity (Danjo et al., 2022; Grolla et al., 2013; Shrivastava et al., 2013).
Taken together with the authors' results showing reduced engulfment of synaptic material by astrocytes in the APP/PS1 model following treatment with the SAM, it’s possible that at least some of the phenotypes they observe are a result of it binding directly to astrocytic mGluR5, which may then subsequently feed back to alter neuronal biology or impact this cell's engulfment phenotype. This may be something worth exploring in future studies.
References:
Haas LT, Salazar SV, Smith LM, Zhao HR, Cox TO, Herber CS, Degnan AP, Balakrishnan A, Macor JE, Albright CF, Strittmatter SM. Silent Allosteric Modulation of mGluR5 Maintains Glutamate Signaling while Rescuing Alzheimer's Mouse Phenotypes. Cell Rep. 2017 Jul 5;20(1):76-88. PubMed.
Delva A, Michiels L, Koole M, Van Laere K, Vandenberghe W. Synaptic Damage and Its Clinical Correlates in People With Early Huntington Disease: A PET Study. Neurology. 2022 Jan 4;98(1):e83-e94. Epub 2021 Oct 18 PubMed.
Onwordi EC, Halff EF, Whitehurst T, Mansur A, Cotel MC, Wells L, Creeney H, Bonsall D, Rogdaki M, Shatalina E, Reis Marques T, Rabiner EA, Gunn RN, Natesan S, Vernon AC, Howes OD. Synaptic density marker SV2A is reduced in schizophrenia patients and unaffected by antipsychotics in rats. Nat Commun. 2020 Jan 14;11(1):246. PubMed.
Danjo Y, Shigetomi E, Hirayama YJ, Kobayashi K, Ishikawa T, Fukazawa Y, Shibata K, Takanashi K, Parajuli B, Shinozaki Y, Kim SK, Nabekura J, Koizumi S. Transient astrocytic mGluR5 expression drives synaptic plasticity and subsequent chronic pain in mice. J Exp Med. 2022 Apr 4;219(4) Epub 2022 Mar 23 PubMed.
Grolla AA, Sim JA, Lim D, Rodriguez JJ, Genazzani AA, Verkhratsky A. Amyloid-β and Alzheimer's disease type pathology differentially affects the calcium signalling toolkit in astrocytes from different brain regions. Cell Death Dis. 2013;4:e623. PubMed.
Shrivastava AN, Kowalewski JM, Renner M, Bousset L, Koulakoff A, Melki R, Giaume C, Triller A. β-amyloid and ATP-induced diffusional trapping of astrocyte and neuronal metabotropic glutamate type-5 receptors. Glia. 2013 Oct;61(10):1673-86. PubMed.
Make a Comment
To make a comment you must login or register.