Can an Antibody Allele Boost Your Alzheimer’s Risk By Giving Herpes an Edge?
Quick Links
The idea that herpes simplex virus type 1 (HSV-1) stokes the fires of Alzheimer’s disease has gained traction in recent years. If true, how would such a common infection trigger neurodegenerative disease in some people, but not others? A study published July 24 in the Journal of Immunology offers a potential explanation. Researchers led by Janardan Pandey of the Medical University of South Carolina and Hugo Lövheim of Umeå University in Sweden report that homozygous carriers of GM17—a variant of the IgG1 antibody gene—have a fourfold greater risk of AD. This common variant, it turns out, is deftly targeted by a decoy receptor HSV-1 expresses. This enables the virus to evade antibody-mediated host responses, i.e., immune defense. The researchers propose that the immune system’s loosened grip on HSV-1 could spur neurodegeneration.
- HSV-1 expresses a decoy receptor that thwarts host antibody responses, especially of the human IgG1 allele GM17.
- Homozygous GM17 carriers had fourfold increased risk of AD.
- GM17 associated with AD risk independently of ApoE or a polygenic risk score.
“These are very interesting results at both the levels of AD genetics and the potential infectious etiology of AD,” commented Rudolph Tanzi and William Eimer, both at Massachusetts General Hospital in Boston. “[The study] starts to address the recurring criticism that the penetrance of HSV-1 infection in the population does not correlate with incidence of AD pathology.”
The majority of the world’s population is infected with HSV-1. Most of the time, the virus is a latent lurker, but it can reactivate throughout life, triggering painful cold sores. The virus can even infect the CNS. Mounting evidence from epidemiological, neuropathological, and cell culture studies have gradually built a case that the virus could promote AD pathogenesis in some people (Jun 2018 news; May 2020 news; and for review, Marcocci et al., 2020).

Common Scourge. HSV-1 virions observed in a transmission electron micrograph. [CDC/Fred Murphy, Wikimedia Commons.]
More than a decade ago, Pandey, an infectious disease immunologist, hypothesized that whether the virus wreaks enough havoc to cause Alzheimer’s in a person could boil down to how well it evades his or her immune response (Pandey 2009). In particular, HSV-1 has evolved tricks to throw antibody-dependent cellular cytotoxicity off its scent. ADCC is an immune mechanism that wipes out infected cells. In ADCC, HSV-1 specific antibodies bind to infected cells with their Fab regions, tagging them for destruction by innate immune cells such as macrophages. These killer cells latch onto their antibody-bound prey via Fcγ receptors (FcγRs). To counteract this process, HSV-1 produces decoy FcγRs, which bind to the antibodies and block their association with effector cells (Frank et al., 1989; Sprague et al., 2006; Lubinski et al., 2011). Importantly, these decoy receptors latch on to a specific variant, also called an allotype, of IgG1 heavy chain—γ marker (GM)17—more strongly than they bind other antibody types (Atherton et al., 2000).
Might their hobbled defense against HSV-1 put homozygous carriers of GM17 at higher risk of AD? To chip away at the answer, Pandey and colleagues genotyped 363 AD cases and 363 age-matched controls for GM17 and GM3, the two possible IgG1 variants at this position in the gene. The samples came from the Northern Sweden Health and Disease Study, a nested case-control study in which samples from people who later developed AD were paired with samples from controls. Lövheim had previously reported that HSV-1 infection synergized with genetic risk factors, including ApoE4, in upping AD risk in this cohort, and Pandey initiated a collaboration with Lövheim to investigate a linkage between GM17 allotype and AD risk (Lopatko Lindman et al., 2019).
In this series, GM17/17 homozygotes were more prevalent among AD cases than among controls, appearing in 19.8 and 10.7 percent of the samples, respectively. The opposite was true for GM3/3 homozygotes, which represented 33.1 percent of cases and 41.3 percent of controls.
The researchers found that GM17/17 came with a fourfold increased risk of AD, independent of ApoE genotype. While ApoE4 and GM17/17 both increased AD risk, the two factors did not appear to synergize to further crank up risk in people who carried both. This held true when the researchers factored in a polygenic risk score— based on genotypes at nine known AD risk loci—that they had previously tied to AD risk.
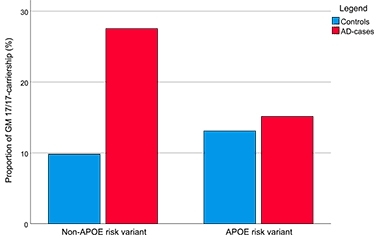
Allotype of AD? Among people unburdened by ApoE4 risk (left), the GM17/17 genotype is more prevalent among AD cases than controls. Among ApoE4 carriers, the proportion of GM17/17 carriers is similar between AD and controls. [Courtesy of Pandey et al., Journal of Immunology, 2020.]
Together, the findings suggest that GM17/17 carriers face higher odds of developing AD. How might this relate to HSV-1’s immune-evasion strategies? The scientists detected anti-HSV-1 IgG antibodies—a proxy for infection with the virus— in about 90 percent of AD cases as well as controls. However, levels of these IgG antibodies were lower in people with two copies of GM17, in keeping with the idea that scavenging by viral decoy receptors enhanced GM17 internalization by infected cells (Ndjamen et al., 2014). While the GM17/17 genotype associated with lower HSV-1-specific IgG antibodies in the serum, ApoE4 carriers had higher levels of IgM antibodies against the virus. IgM antibodies predominate at the beginning of an immune response, hence their presence suggests a recent reactivation of the virus. Taken together, the findings suggest to the authors that GM17/17 and ApoE may influence immunity against HSV-1 via distinct mechanisms, both of which could potentially sway a person’s Alzheimer’s risk.
Lövheim told Alzforum that while HSV-1 immunoevasion is the most likely explanation for the link between GM17 and AD risk, it’s possible that the allele works via other mechanisms. For example, GM alleles could differentially influence antibody-mediated uptake of Aβ by microglia. Tanzi and Eimer noted that HSV-1 is not the only pathogen that has an FcγR decoy up its sleeves. “Human cytomegalovirus glycoprotein 34 and Staphylococcus aureus protein A both promote immune suppression during infection and bind to the same FcγR, suggesting that other pathogens should also be explored for their potential roles in AD etiology,” they wrote.
Even if HSV-1 is the culprit, the data do not prove that a weakened antibody response against the virus would promote its entry into the brain and/or influence AD pathogenesis. The authors proposed a deeper investigation into potential mechanisms, and that the genetic studies should be replicated in large, multiethnic cohorts.
Why hasn’t GM17 popped up in larger genome-wide association studies before? IgG genes are multiallelic and highly homologous, making them difficult to genotype. As such, single-nucleotide polymorphisms that distinguish between GM3 and GM17 are not included on the genotyping chips used in AD GWAS. AD geneticists contacted by Alzforum confirmed that the SNP in question— rs1071803—was not included in the largest recent AD GWAS (Calonga-Solís et al., 2019; Jansen et al., 2019). However, they also noted that another nearby SNP—rs12147642— is in high linkage disequilibrium with the GM3,17 SNP, and it was not tied to AD risk. According to an NIH LD pairing tool, the two SNPs have a 94 percent chance of co-inheritance (R2=0.94). The authors noted the possibility that other GM allotypes in high linkage disequilibrium with the GM3,17 alleles, such as GM1 and GM21 alleles within the IGHG1 and IGHG3 genes, respectively, may have been responsible for the link to AD risk.
GM3,17 alleles of IgG1 have not been directly tested in any AD GWAS. That said, one large GWAS has tied variants near the immunoglobulin heavy chain variable (IGHV) locus to the disease (Witoelar et al., 2018). Rare variants in the IGHG3 gene, which encodes the constant region of IgG3, have emerged in whole-exome sequencing studies (Aug 2018 news).—Jessica Shugart
References
News Citations
- Herpes Triggers Amyloid—Could This Virus Fuel Alzheimer’s?
- Herpes Simplex Virus Triggers Amyloidosis in 3D Neural Cultures
- Largest AD Whole-Exome Study to Date Finds Two New Risk Genes
Paper Citations
- Marcocci ME, Napoletani G, Protto V, Kolesova O, Piacentini R, Li Puma DD, Lomonte P, Grassi C, Palamara AT, De Chiara G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020 Oct;28(10):808-820. Epub 2020 May 5 PubMed.
- Pandey JP. Immunoglobulin GM genes as functional risk and protective factors for the development of Alzheimer's disease. J Alzheimers Dis. 2009;17(4):753-6. PubMed.
- Frank I, Friedman HM. A novel function of the herpes simplex virus type 1 Fc receptor: participation in bipolar bridging of antiviral immunoglobulin G. J Virol. 1989 Nov;63(11):4479-88. PubMed.
- Sprague ER, Wang C, Baker D, Bjorkman PJ. Crystal structure of the HSV-1 Fc receptor bound to Fc reveals a mechanism for antibody bipolar bridging. PLoS Biol. 2006 Jun;4(6):e148. Epub 2006 May 2 PubMed.
- Lubinski JM, Lazear HM, Awasthi S, Wang F, Friedman HM. The herpes simplex virus 1 IgG fc receptor blocks antibody-mediated complement activation and antibody-dependent cellular cytotoxicity in vivo. J Virol. 2011 Apr;85(7):3239-49. Epub 2011 Jan 12 PubMed.
- Atherton A, Armour KL, Bell S, Minson AC, Clark MR. The herpes simplex virus type 1 Fc receptor discriminates between IgG1 allotypes. Eur J Immunol. 2000 Sep;30(9):2540-7. PubMed.
- Lopatko Lindman K, Weidung B, Olsson J, Josefsson M, Kok E, Johansson A, Eriksson S, Hallmans G, Elgh F, Lövheim H. A genetic signature including apolipoprotein Eε4 potentiates the risk of herpes simplex-associated Alzheimer's disease. Alzheimers Dement (N Y). 2019;5:697-704. Epub 2019 Nov 4 PubMed.
- Ndjamen B, Farley AH, Lee T, Fraser SE, Bjorkman PJ. The herpes virus Fc receptor gE-gI mediates antibody bipolar bridging to clear viral antigens from the cell surface. PLoS Pathog. 2014 Mar;10(3):e1003961. Epub 2014 Mar 6 PubMed.
- Calonga-Solís V, Malheiros D, Beltrame MH, Vargas LB, Dourado RM, Issler HC, Wassem R, Petzl-Erler ML, Augusto DG. Unveiling the Diversity of Immunoglobulin Heavy Constant Gamma (IGHG) Gene Segments in Brazilian Populations Reveals 28 Novel Alleles and Evidence of Gene Conversion and Natural Selection. Front Immunol. 2019;10:1161. Epub 2019 Jun 4 PubMed.
- Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hägg S, Athanasiu L, Voyle N, Proitsi P, Witoelar A, Stringer S, Aarsland D, Almdahl IS, Andersen F, Bergh S, Bettella F, Bjornsson S, Brækhus A, Bråthen G, de Leeuw C, Desikan RS, Djurovic S, Dumitrescu L, Fladby T, Hohman TJ, Jonsson PV, Kiddle SJ, Rongve A, Saltvedt I, Sando SB, Selbæk G, Shoai M, Skene NG, Snaedal J, Stordal E, Ulstein ID, Wang Y, White LR, Hardy J, Hjerling-Leffler J, Sullivan PF, van der Flier WM, Dobson R, Davis LK, Stefansson H, Stefansson K, Pedersen NL, Ripke S, Andreassen OA, Posthuma D. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019 Mar;51(3):404-413. Epub 2019 Jan 7 PubMed.
- Witoelar A, Rongve A, Almdahl IS, Ulstein ID, Engvig A, White LR, Selbæk G, Stordal E, Andersen F, Brækhus A, Saltvedt I, Engedal K, Hughes T, Bergh S, Bråthen G, Bogdanovic N, Bettella F, Wang Y, Athanasiu L, Bahrami S, Le Hellard S, Giddaluru S, Dale AM, Sando SB, Steinberg S, Stefansson H, Snaedal J, Desikan RS, Stefansson K, Aarsland D, Djurovic S, Fladby T, Andreassen OA. Meta-analysis of Alzheimer's disease on 9,751 samples from Norway and IGAP study identifies four risk loci. Sci Rep. 2018 Dec 27;8(1):18088. PubMed.
External Citations
Further Reading
News
Webinars
Primary Papers
- Pandey JP, Olsson J, Weidung B, Kothera RT, Johansson A, Eriksson S, Hallmans G, Elgh F, Lövheim H. An Ig γ Marker Genotype Is a Strong Risk Factor for Alzheimer Disease, Independent of Apolipoprotein E ε4 Genotype. J Immunol. 2020 Sep 1;205(5):1318-1322. Epub 2020 Jul 24 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Massachusetts General Hospital
Massachusetts General Hospital, Harvard
In this study, Pandey et al. report that a γ marker (GM) allotype more susceptible to HSV-1 immune evasion mechanisms is present at higher rates in subjects with Alzheimer’s disease. The authors show that carriers homozygous for the GM 17 allotype were significantly more present among AD cases than controls. The fourfold increased risk of AD in these carriers was independent of APOE risk.
These are very interesting results, at the levels of both AD genetics and the potential infectious etiology of AD. The IGHV locus has been previously implicated to be associated with AD by GWAS, adding to the strength of the study. It is also worth noting that other viral-infection-related genes have been previously associated with AD, including PILRA and ITGB3
GM 17’s significantly increased affinity for HSV1’s gE-gI complex, presenting a more effective immune modulation by the virus, is an intriguing mechanism for a potential viral etiology of AD, but not unique to HSV-1. Human cytomegalovirus glycoprotein 34 and Staphylococcus aureus protein A both promote immune suppression during infection and bind to the same Fcγ, suggesting that other pathogens should also be explored for their potential roles in AD etiology.
So far, we do not have a smoking gun for a potential viral etiology of AD. But many of us are actively exploring it. Pandey et al. presents a case for the role of HSV-1 in AD and starts to address the recurring criticism that the penetrance of HSV-1 infection in the population does not correlate with the incidence of AD pathology.
Icahn School of Medicine
The coronavirus pandemic has placed in stark relief our primitive understanding of virology and the complications caused by viral infection because of immunological, immunogenetic, inflammatory, coagulopathic, and vasculopathic events. This an especially apt time for the neurodegenerative disease field to revisit the longstanding but still controversial implication that herpesviruses may sometimes play some roles in the pathogenesis of AD. A recent "big data" paper by a multi-lab collaborative group (Readhead et al., 2018) reignited the focus on herpes viruses in AD.
A related area of investigation was highlighted in this Alzforum story. Pandey and colleagues focus on evidence that allotypes of immune-inflammatory system components may play roles in AD risk. The GM17 allotype of IgG1, linked in this paper to AD, has a history of specifying host response to a variety of viral infections.
Another entirely different virus-neurodegeneration story was recently reported (Dembny et al., 2020). These investigators demonstrated that retroviral proteinase HERVK RNA can apparently cause neurodegeneration through activation of Toll-like receptors. This mechanism is invoked as a potential explanation for reports that HERVK plays a role in ALS (Li et al., 2015). The notion that an RNA can be neuroactive dovetails well with other evidence that RNAs and microRNAs can play either pathogenic or protective roles in the pathogenesis of a range of neurodegenerative diseases (e.g., miR155 in Readhead et al., 2020).
A key question is whether the new data implicating viruses might be actionable. To investigate that possibility, Gold et al. (2019) conducted a clinical trial in ALS using a cocktail of multiple antiviral drugs. That trial generated sufficiently convincing clinical and biomarker evidence for safety and benefit that a large international trial of the same cocktail is now being planned. Devanand et al. (2020) have set out along a similar path in their trial of valacyclovir in AD.
Obviously, the current acute public health emergency calls for advancing our understanding of viruses and the pleiotropic havoc they can wreak, both acutely and chronically. One hopes that the intense current interest in virology and immunology will generate new information and tools that will enable us to clarify not only the multi-organ pathogenesis of SARS-CoV2 disease, but also the neuropathogenesis that potentially links herpesviruses to AD.
References:
Dembny P, Newman AG, Singh M, Hinz M, Szczepek M, Krüger C, Adalbert R, Dzaye O, Trimbuch T, Wallach T, Kleinau G, Derkow K, Richard BC, Schipke C, Scheidereit C, Stachelscheid H, Golenbock D, Peters O, Coleman M, Heppner FL, Scheerer P, Tarabykin V, Ruprecht K, Izsvák Z, Mayer J, Lehnardt S. Human endogenous retrovirus HERV-K(HML-2) RNA causes neurodegeneration through Toll-like receptors. JCI Insight. 2020 Apr 9;5(7) PubMed.
Devanand DP, Andrews H, Kreisl WC, Razlighi Q, Gershon A, Stern Y, Mintz A, Wisniewski T, Acosta E, Pollina J, Katsikoumbas M, Bell KL, Pelton GH, Deliyannides D, Prasad KM, Huey ED. Antiviral therapy: Valacyclovir Treatment of Alzheimer's Disease (VALAD) Trial: protocol for a randomised, double-blind,placebo-controlled, treatment trial. BMJ Open. 2020 Feb 6;10(2):e032112. PubMed.
Gold J, Rowe DB, Kiernan MC, Vucic S, Mathers S, van Eijk RP, Nath A, Garcia Montojo M, Norato G, Santamaria UA, Rogers ML, Malaspina A, Lombardi V, Mehta PR, Westeneng HJ, van den Berg LH, Al-Chalabi A. Safety and tolerability of Triumeq in amyotrophic lateral sclerosis: the Lighthouse trial. Amyotroph Lateral Scler Frontotemporal Degener. 2019 Nov;20(7-8):595-604. Epub 2019 Jul 8 PubMed.
Li W, Lee MH, Henderson L, Tyagi R, Bachani M, Steiner J, Campanac E, Hoffman DA, von Geldern G, Johnson K, Maric D, Morris HD, Lentz M, Pak K, Mammen A, Ostrow L, Rothstein J, Nath A. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med. 2015 Sep 30;7(307):307ra153. PubMed.
Readhead B, Haure-Mirande JV, Funk CC, Richards MA, Shannon P, Haroutunian V, Sano M, Liang WS, Beckmann ND, Price ND, Reiman EM, Schadt EE, Ehrlich ME, Gandy S, Dudley JT. Multiscale Analysis of Independent Alzheimer's Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron. 2018 Jul 11;99(1):64-82.e7. Epub 2018 Jun 21 PubMed.
Readhead B, Haure-Mirande JV, Mastroeni D, Audrain M, Fanutza T, Kim SH, Blitzer RD, Gandy S, Dudley JT, Ehrlich ME. miR155 regulation of behavior, neuropathology, and cortical transcriptomics in Alzheimer's disease. Acta Neuropathol. 2020 Sep;140(3):295-315. Epub 2020 Jul 14 PubMed.
It's heartwarming to see increased openness to the relatively unexplored possibilities that microbes can trigger Alzheimer's. I hope in this rush to pinpoint intracellular and genomic influence, we will still urge epidemiologists to further investigate possible macro clues, such as reports that neurosurgeons and caregivers of Alzheimer's patients develop more Alzheimer's disease than statistics might predict.
Make a Comment
To make a comment you must login or register.