Could Silencing Neuroligin-1 Drive Synaptic Loss in AD?
Quick Links
The hooks and loops on new Velcro hold tight at first, but if one side gets damaged, the connection falls apart. A new study suggests that something similar happens to synapses in Alzheimer's disease. Scientists led by Mohamed Naguib, Cleveland Clinic, Ohio, found that through a complex series of steps, Aβ fibrils suppress a cell-adhesion protein called neuroligin-1, a postsynaptic protein that binds presynaptic neurexins. Loss of neuroligin ablated dendritic spines, disrupted long-term potentiation, and caused memory deficits in rats. Activated microglia appeared to kick-start the whole process by silencing the neuroligin-1 gene. The results, which connect several mechanisms independently implicated in Alzheimer's disease, appeared online January 19 in Nature Neuroscience.
"At the molecular level, these authors provide new insight into how Aβ-induced glial activation and its inflammation has an impact on the epigenetic landscape," said Li-Huei Tsai of Massachusetts Institute of Technology, Cambridge, who was not involved in the project. The work adds to mounting evidence that inflammation is a negative player in AD, she said.
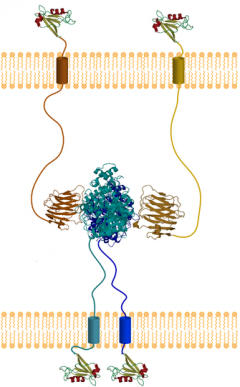
Neuroligins connect synapses, but may be in short supply in Alzheimer's. Image courtesy of Studentne/WikiMedia Commons
Neuroligin-1 in the post-synaptic membrane links up with neurexin partners on the pre-synaptic side to form and shore up excitatory connections. Because γ-secretase cleaves both these type-1 membrane proteins in addition to amyloid precursor protein, researchers have wondered if the cell adhesion molecules might be involved in AD pathology. Mutations in neuroligin have been tied to autism (see Cornoletti et al., 2004); however, no variants in the gene have been associated with AD, and a genome-wide association study turned up a protective allele only in neurexin 3 (see Martinez-Mir et al., 2013). However, given previous indications that neuroligin may be important for spatial memory (see Blundell et al., 2010), Naguib and colleagues had a hunch that it might play a role in AD, too. Naguib runs an anesthesiology lab and in recent years has been studying the role of microglia in neuropathy and other neuroinflammatory conditions, including AD. His group wanted to explore whether microglial activation affects neuroligin.
To start out, the researchers tested if amyloid suppressed neuroligin levels. Co-lead authors Bihua Bie and Jiang Wu injected Aβ40 fibrils into the hippocampal CA1 area of rats. This caused memory deficits. It also reduced excitatory transmission and weakened long-term potentiation in hippocampal slices. In these samples, neuroligin-1 and its mRNA dwindled. To figure out why, the researchers looked at the epigenetic regulation of neuroligin-1 and discovered that acetylation had fallen in the gene's promoter region. Because acetylation opens up DNA to transcription, the finding suggested neuroligin-1 was being silenced.
In support of this, the researchers found more histone deacetylase-2 (HDAC2) in the neuroligin promoter region in the Aβ40-treated hippocampal slices compared to controls. Here, DNA also boasted fewer acetyl and more methyl groups, typically a recipe for silencing genes. In APPSwe/PSEN1dE9 mice, which accumulate amyloid fibrils, the neuroligin promoter region bound more HDAC2/MeCP2 complexes and made less neuroligin compared to control mice.
Methyl-CpG binding protein 2 (MeCP2) binds methylated DNA and either represses or activates gene transcription. Mutations in MeCP2 have been tied to Rett syndrome, while MeCP2 loss- or gain-of-function impairs synaptic plasticity in animal models (for a review, see Na et al., 2013).
Where did microglia come into play? These cells likely touched off the whole process, the authors wrote. Injecting lipopolysaccharide (LPS) into the CA1 activated microglia and mirrored the effects of the amyloid fibrils. On the other hand, when the researchers prevented microglial activation by pairing minocycline and Aβ fibril injections, the mice maintained normal neuroligin-1 expression, synapse number, and learning and memory.
The results suggest that microglial inflammation reduces neuroligin and plays a role in amyloid-induced damage, wrote the authors. A previous study also found that minocycline mitigated the LTP-dampening effects of Aβ on slices of the dentate gyrus from rodents, but attributed this effect to a reduction in nitric oxide (see Jul 2004 news story).
Scientists not involved in the work thought it interesting, but cautioned that it will need to be replicated and further explored. Tsai said the study is thoroughly done and that it is consistent with research from her lab suggesting that Aβ boosts HDAC2 levels and that overactive HDAC2 suppresses memory in mice (see Gräff et al., 2012; May 2009 news story). However, because it is nearly impossible for an epigenetic mechanism to influence just one gene, other genes are likely involved in amyloid-induced synaptic dysfunction, she added.
Azusa Shiohara and Taisuke Tomita, University of Tokyo, wondered by how much neuroligin must drop to disrupt synapses, and how levels of the protein in AD patients compare with those of healthy controls. If the neuroligin finding pans out, the mechanisms that govern this protein's cellular concentration could be targeted for AD treatments, they wrote to Alzforum (see full comment below). Michael Heneka, University of Bonn, Germany, noted that inflammation likely harms additional aspects of synaptic health, aside from neuroligin (see full comment below).
Frank Heppner, Charité–Universitätsmedizin, Berlin, wrote that Aβ40 fibrils constitute a narrow slice of the full spectrum of Aβ species found in AD brains, and suggested these findings be replicated with a more representative model. What's more, injection of Aβ mimics acute injury with short-term exposure of microglia to the amyloid, whereas amyloid exposure in AD is chronic. It could cause a different inflammatory response altogether, Heppner wrote (see full comment below). Naguib pointed out that the group saw similar results in the transgenic mouse model, which models chronic exposure to Aβ.
Naguib said his next step is to clinically test a drug, called MDA7, which his group helped develop for neuroinflammatory conditions. MDA7 is an agonist for the microglial cannabinoid type-2 (CB2) receptor, which inhibits release of proinflammatory molecules. Naguib and colleagues reported that MDA7 treated neuropathic pain in rats (see Naguib et al., 2008) and reduced synaptic dysfunction and cognitive impairment that accompanied Aβ40 injections (see Wu et al., 2013). Intervening in the pathway leading from microglia to synapse damage could treat multiple neuroinflammatory diseases, he told Alzforum.—Gwyneth Dickey Zakaib
References
News Citations
- Microglia Aid and Abet Aβ Toxicity and Clearance
- It’s an HDAC2 Wrap— Memory-suppressing DNA Modifier Identified
Paper Citations
- Comoletti D, De Jaco A, Jennings LL, Flynn RE, Gaietta G, Tsigelny I, Ellisman MH, Taylor P. The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J Neurosci. 2004 May 19;24(20):4889-93. PubMed.
- Martinez-Mir A, González-Pérez A, Gayán J, Antúnez C, Marín J, Boada M, Lopez-Arrieta JM, Fernández E, Ramírez-Lorca R, Sáez ME, Ruiz A, Scholl FG, Real LM. Genetic study of neurexin and neuroligin genes in Alzheimer's disease. J Alzheimers Dis. 2013 Jan 1;35(2):403-12. PubMed.
- Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C, Bolliger MF, Südhof TC, Powell CM. Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J Neurosci. 2010 Feb 10;30(6):2115-29. PubMed.
- Na ES, Nelson ED, Kavalali ET, Monteggia LM. The impact of MeCP2 loss- or gain-of-function on synaptic plasticity. Neuropsychopharmacology. 2013 Jan;38(1):212-9. Epub 2012 Jul 11 PubMed.
- Gräff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, Su SC, Samiei A, Joseph N, Haggarty SJ, Delalle I, Tsai LH. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012 Mar 8;483(7388):222-6. PubMed.
- Naguib M, Diaz P, Xu JJ, Astruc-Diaz F, Craig S, Vivas-Mejia P, Brown DL. MDA7: a novel selective agonist for CB2 receptors that prevents allodynia in rat neuropathic pain models. Br J Pharmacol. 2008 Dec;155(7):1104-16. Epub 2008 Sep 1 PubMed.
- Wu J, Bie B, Yang H, Xu JJ, Brown DL, Naguib M. Activation of the CB(2) receptor system reverses amyloid-induced memory deficiency. Neurobiol Aging. 2012 Jul 12; PubMed.
Other Citations
Further Reading
Papers
- Sindi IA, Tannenberg RK, Dodd PR. A role for the neurexin-neuroligin complex in Alzheimer's disease. Neurobiol Aging. 2014 Apr;35(4):746-56. Epub 2013 Nov 6 PubMed.
- Martinez-Mir A, González-Pérez A, Gayán J, Antúnez C, Marín J, Boada M, Lopez-Arrieta JM, Fernández E, Ramírez-Lorca R, Sáez ME, Ruiz A, Scholl FG, Real LM. Genetic study of neurexin and neuroligin genes in Alzheimer's disease. J Alzheimers Dis. 2013 Jan 1;35(2):403-12. PubMed.
- Dinamarca MC, Weinstein D, Monasterio O, Inestrosa NC. The synaptic protein neuroligin-1 interacts with the amyloid β-peptide. Is there a role in Alzheimer's disease?. Biochemistry. 2011 Sep 27;50(38):8127-37. PubMed.
News
Primary Papers
- Bie B, Wu J, Yang H, Xu JJ, Brown DL, Naguib M. Epigenetic suppression of neuroligin 1 underlies amyloid-induced memory deficiency. Nat Neurosci. 2014 Feb;17(2):223-31. Epub 2014 Jan 19 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Charité Universitaetsmedizin Berlin
This study by Bie and colleagues is certainly very interesting. It nicely links microglia-mediated inflammation in general, i.e., irrespective of the cause/trigger of microglia activation, to epigenetic changes and subsequent phenotypes, including memory changes. Thus it brings yet another strong piece of evidence that neuroinflammation and microglia action impact neuronal function. However, the injection of synthetic Aβ peptides (apparently restricted to Aβ 1-40) does not represent the full picture and spectrum of Aβ and amyloid species found in AD. Thus, the described phenotype and mechanistic explanation needs to be confirmed at least in a transgenic AD model of cerebral amyloidosis and/or by using amyloid fibrils isolated from human or transgenic mouse AD brain. Moreover, this type of injection model rather mimics an acute injury, and the microglia only have short-term exposure to the Aβ, while the chronic and progressive nature of AD, even in mouse models, likely induces a different inflammatory response.
In conclusion, I think these are very interesting and—in general—convincing findings. However, they describe a mechanism that may not necessarily account for AD, or at least needs to be tested more stringently in an AD context.
View all comments by Frank HeppnerKlinik und Poliklinik für Neurologie
Chronic neuroinflammation is a common feature of several degenerative disorders of the central nervous system, including amyotrophic lateral sclerosis, Parkinson´s disease, and Alzheimer´s disease. It is widely assumed that the sustained presence of inflammatory cytokines, chemokines, or radical oxygen species contribute to neuronal dysfunction and demise in these neurodegenerative diseases. Recent evidence suggests that microglial activation affects synaptic morphology and plasticity in AD. Precisely how inflammatory mediators affect neuronal function and morphology remains largely unknown.
The present paper by Bie and colleagues investigates the effect of a direct injection of either fibrillar Aβ1-40 or bacterial lipopolysaccharide into the rat hippocampus, thereby modeling acute toll-like receptor 4 mediated brain inflammation. A key finding of this study shows that Aβ1-40 and LPS induce an upregulation of HDAC2 activity, which in turn leads to the suppression of neuroligin1. Neuroligin1, a postsynaptic protein, forms a complex with the presynaptic protein neurexin and this trans-synaptic complex contributes to synaptic plasticity and efficacy. Consequently, neuroligin1 reduction associated with suppression of long-term potentiation and impaired spatial memory tested by the Morris Water Maze paradigm. A cofactor for the HDAC2 induced transcriptional suppression of neuroligin1 is Mecp2, since administration of its siRNA decreased HDAC2 occupancy in the neuroligin- promoter region. Both Aβ1-40 and LPS injection caused acute microglial activation with an increase of CD11b positivity and interleukin-1β in the rat hippocampus. Microinjection of minocycline decreased both CD11b and interleukin-1β levels and restored neuroligin-1 mRNA and protein levels, thereby improving synapse function and spatial memory.
This paper is important since it sheds further light on the mechanisms by which inflammatory molecules affect neuronal functions relevant for memory formation. It underlines that Aβ peptides, at least on an in-vivo level, may not affect synaptic function alone, but act through the release of inflammatory molecules by activated microglia. It stands in line with previous results showing that Aβ-evoked inflammation suppresses LTP also through the release of nitric oxide by NOS2 (Wang et al., 2004). Nevertheless, inflammatory mediators may affect more than one system relevant for synaptic functioning as indicated by a recent paper (see Tong et al., 2012). While the present mechanisms require further functional studies in models of chronic Aβ stimulation, the protective effect on innate immune blockade has already been shown in such models (Kummer et al., 2011; Heneka et al., 2013).
References:
Wang Q, Rowan MJ, Anwyl R. Beta-amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J Neurosci. 2004 Jul 7;24(27):6049-56. PubMed.
Tong L, Prieto GA, Kramár EA, Smith ED, Cribbs DH, Lynch G, Cotman CW. Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1β via p38 mitogen-activated protein kinase. J Neurosci. 2012 Dec 5;32(49):17714-24. PubMed.
Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, Walter J, Pape HC, König S, Roeber S, Jessen F, Klockgether T, Korte M, Heneka MT. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron. 2011 Sep 8;71(5):833-44. PubMed.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013 Jan 31;493(7434):674-8. Epub 2012 Dec 19 PubMed.
View all comments by Michael HenekaThe University of Tokyo
University of Tokyo
Several researchers have reported that Aβ fibrils affect synaptic plasticity, learning and memory in-vitro and in-vivo. However the precise mechanism of synaptic toxicity of Aβ remains elusive. Neuroligin 1 (NLG1) is one of the postsynaptic adhesion molecules bridging the synaptic cleft with its presynaptic ligand, neurexin. NLG1 has been implicated in the formation and stabilization of central glutamatergic synapses. Genetic studies revealed that NLG1 plays critical roles in the etiology of autism spectrum disorder and neuropsychiatric diseases. However, the pathophysiological role of NLG1 in Alzheimer disease still remains unknown.
Bie et al. analyzed the Aβ-injected rat as the amyloid-induced memory deficiency model, and found that amyloid fibrils caused the synaptic dysfunction via epigenetic changes in the expression of NLG1. They also revealed that these changes are mediated through neuroinflammation pathway. The results presented here imply that the epigenetic mechanism links amyloid-induced neuroinflammation and synaptic dysfunction through the regulation of NLG1 expression.
It would be interesting to study how much reduction in NLG1 expression is sufficient for the synaptic dysfunction. As NLG1 levels are regulated by epigenetic (this study), translational (Gkogkas et al., 2013), and proteolytic (Suzuki et al., 2012; Peixoto et al., 2012) mechanisms, pharmacological modulation of these pathways might have a potential as anti-memory deficiency therapeutics. Also, it would be intriguing to analyze whether the other molecules regulated by MeCP2/HDAC2 complex are involved in the Aβ toxicity. In addition, analysis of NLG1 levels in the brains of preclinical AD, MCI and AD patients may provide an important information when and where the synaptic dysfunction by Aβ starts and spreads. In sum, this study supports the notion that the alterations in the synaptic adhesion molecules by amyloid fibrils or other insults play pathological roles in Alzheimer disease and neurodegenerative disease by disruption of connectome in human brains.
References:
Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, Major F, Lasko P, Ruggero D, Nader K, Lacaille JC, Sonenberg N. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2013 Jan 17;493(7432):371-7. Epub 2012 Nov 21 PubMed.
Suzuki K, Hayashi Y, Nakahara S, Kumazaki H, Prox J, Horiuchi K, Zeng M, Tanimura S, Nishiyama Y, Osawa S, Sehara-Fujisawa A, Saftig P, Yokoshima S, Fukuyama T, Matsuki N, Koyama R, Tomita T, Iwatsubo T. Activity-dependent proteolytic cleavage of neuroligin-1. Neuron. 2012 Oct 18;76(2):410-22. PubMed.
Peixoto RT, Kunz PA, Kwon H, Mabb AM, Sabatini BL, Philpot BD, Ehlers MD. Transsynaptic signaling by activity-dependent cleavage of neuroligin-1. Neuron. 2012 Oct 18;76(2):396-409. PubMed.
View all comments by Azusa ShioharaMake a Comment
To make a comment you must login or register.