CRISPR Screens Net Tau Partners in Human Neurons, Brain
Quick Links
In tauopathies, tau forms aggregates in specific areas of the brain, but it is unclear how. Two new studies suggest that in induced pluripotent stem cell-derived neurons, at least, thousands of proteins may modulate the process. Scientists led by Martin Kampmann at the University of California, San Francisco, used CRISPR interference to screen all protein-coding genes for those that influence oligomerization of the V337M tau mutant. In a bioRxiv preprint uploaded June 26, they reported that 1,150 genes fit the bill, including those involved in autophagy, mitochondrial function, the proteasome, and a process called UFMylation. In another bioRxiv preprint uploaded June 22, researchers led by Li Gan at Weill Cornell Medicine in New York, who collaborated with Kampmann, reported a similar CRISPRi screen in iPSC-derived neurons carrying the P301S tau mutant. They identified 500 genes, with top hits falling in retromer, mitochondrial, and UFMylation pathways. Both studies pegged the dearth of two UFMylation genes, UFM1 and UFL1, to fewer aggregates.
- CRISPRi screens peg proteins that influence the formation of tau oligomers.
- Among them are mitochondrial proteins that limit reactive oxygen species.
- ROS weaken the proteasome, which churns out tau fragments that oligomerize.
- Inhibiting UFMylation, a ubiquitinylation-like process, reduced tau aggregation.
Marc Diamond of the University of Texas, Southwestern Medical Center, Dallas, noted that the data shows the power of CRISPRi to pick out genes with subtle effects on cell biology, though he thought further corroboration was required. “I would need a little more data to convince me that they have truly found modifiers of tau oligomer formation/clearance,” he wrote (comment below).
In Kampmann’s lab, first author Avi Samelson and colleagues used CRISPRi to systematically knock down about 21,000 protein-coding genes in iPSC-derived human neurons carrying three-repeat (3R) V337M tau, a mutation that causes frontotemporal dementia. Samelson then measured tau oligomers using three antibodies that preferentially bind small aggregates over tau monomers or fibrils: T22, TOC1, and M204 (Nov 2010 conference news; Ward et al., 2013; Abskharon et al., 2020). Sorting the cells into those with the least and most oligomers allowed the researchers to determine which genes influenced tau aggregation.
Of the 21,000 genes knocked down, 1,143 did so (see image below). “As a scientist who works mainly with purified macromolecules, I was surprised to learn that such a large number of cellular factors affect the aggregation of tau,” wrote co-author David Eisenberg of the University of California, Los Angeles.

Tau Modulators. Some genes silenced by CRISPRi lead to fewer (left) or more (right) tau oligomers than average. Colors correspond to molecular pathways, such as mitochondrial function (red), the ubiquitin/proteasome system (purple), and autophagy (green). [Courtesy of Samelson et al., bioRxiv, 2023.]
Among the top hits were MAPT itself, as well as genes that regulate autophagy and methyl-6-adenosine (m6A) formation, processes known to alter tau oligomerization (Silva et al., 2020; Sep 2021 news). Knocking down MAPT and m6A genes reined in tau oligomers while stifling autophagy had the opposite effect.
Other top genes fell into pathways that had not been directly linked to tau aggregation before. Mitochondrial function strongly influenced oligomer formation. Knocking down genes in the electron transport chain, for example, increased oligomer levels. Because suppressing these genes increases the amount of reactive oxygen species (ROS) in neurons, the authors speculated that oxidation of tau might kickstart oligomerization.
Indeed, when the researchers treated neurons with hydrogen peroxide or electron transport chain inhibitors, which boost ROS, levels of tau oligomers, specifically a 25 kDa N-terminal tau fragment, rose inside the neurons. A version of tau that replaced methionine residues, which are highly susceptible to oxidation, with leucines, which cannot be oxidized, formed much less of this fragment. Inhibiting the proteasome also dialed down the peptide, suggesting that this protein degradation complex makes it. Kampmann thinks that disruption of mitochondria creates ROS that oxidize tau, triggering the proteasome to misprocess it into aggregate-prone oligomers. “A decrease in mitochondrial function is a hallmark of aging, so it could help explain why tau aggregation depends on age,” he told Alzforum.
The scientists believe the peptide in these oligomers spans amino acids 1 to 200 in three-repeat tau, or up to residue 250 in four-repeat tau. Similar fragments of tau find their way into the cerebrospinal fluid early in AD, namely N-terminal peptides phosphorylated at residues 181 or 231. “These biomarkers could be a signature of oxidative stress-induced proteasome dysfunction,” the authors suggested. Diamond wasn’t so sure. “Without carefully comparing the identity of the fragments created by stress to those detected as biomarkers in patients, it is a stretch to suggest that this is the cause of the biomarkers,” he wrote. The authors did find that the 25 kDa fragment was released by neurons into the cell culture medium, suggesting that in the brain it might make its way into the CSF.
The top hits in the CRISPRi screen also comprised proteins of the ubiquitin/proteasome system. Knockdown of these proteins, including chaperones, deubiquitinases, and ubiquitin ligases, flooded cells with oligomers. One such ligase stood out. Neurons deficient in cullin 5 were particularly packed with oligomers, and in immunoprecipitation experiments, the ligase bound tau. Kampmann had previously found that in the AD brain, neurons resilient to neurofibrillary tangles upregulated the CUL5 gene (Leng et al., 2021).
Two lesser-known processes also had a hand in tau oligomerization: the synthesis of glycosylphosphatidylinositol, a lipid that anchors proteins to membranes, and UFMylation. A post-translational modification similar to ubiquitination, the latter adds the ubiquitin-fold modifier 1 (UFM1) peptide to ribosomal subunits that have stalled during translation, tagging their nascent proteins for degradation. UFMylation also spurs autophagy of the ribosome-studded endoplasmic reticulum when that organelle is damaged (Komatsu et al., 2004; Wang et al., 2020; Liang et al., 2020). Silencing either process tempered oligomer load.
Scientists in Gan’s lab also found that inhibiting UFMylation suppressed tau aggregation. First author Celeste Parra Bravo took the top 1,073 hits identified by Kampmann’s lab, and rescreened them using CRISPRi in iPSC-derived neurons engineered to express 4R tau with the P301S mutation, which also causes FTD. She then sorted neurons by pathogenic tau load, as judged by the MC1 antibody. About half of the genes tested modulated aggregation. Knocking down genes involved in mitochondrial function and UFMylation suppressed tau aggregation, while curbing retromer function and endolysosomal trafficking genes encouraged it (see image below).
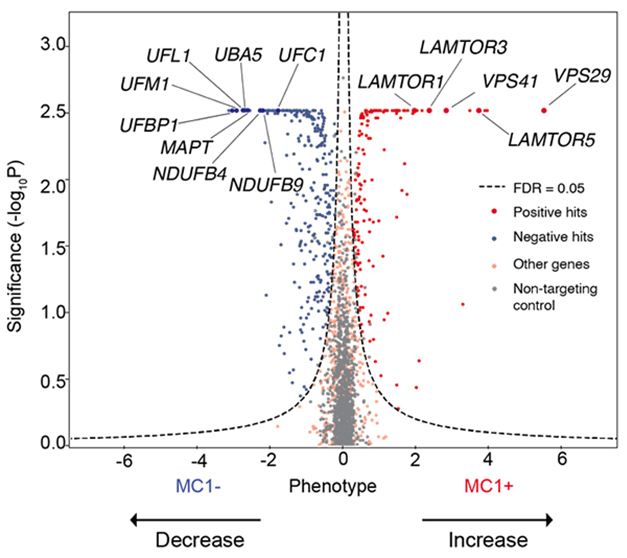
Aggregation Modifiers. A CRISPRi screen in 4R-tau neurons detected genes that, when deleted, led to more (right) or less (left) pathological tau. Genes code proteins involved in UFMylation (UFM1, UFL1, UBA5, UFC1, UFBP1) and mitochondrial function (NDUFB4, NDUFB9). [Courtesy of Parra Bravo et al., bioRxiv, 2023.]
Curious about the UFMylation, Parra Bravo tested iPSC-derived neurons and neurons isolated from postmortem AD cortical tissue for proteins that help drive the process. She detected less UFM1 in cells harboring tau tangles. The scientists speculated that a lack of UFM1 might cause a buildup of protein debris at the ER, triggering ER stress and downstream protein aggregation. “These findings provide the first evidence of involvement of UFMylation in tauopathy and that targeting UFMylation could be beneficial in reducing tau propagation,” they wrote.—Chelsea Weidman Burke
References
Research Models Citations
News Citations
- San Diego: Tau Oligomer Antibodies Relieve Motor Deficits in Mice
- Methylated RNA: A New Player in Tau Toxicity?
Mutations Citations
Paper Citations
- Ward SM, Himmelstein DS, Lancia JK, Fu Y, Patterson KR, Binder LI. TOC1: Characterization of a Selective Oligomeric Tau Antibody. J Alzheimers Dis. 2013;37(3):593-602. PubMed.
- Abskharon R, Seidler PM, Sawaya MR, Cascio D, Yang TP, Philipp S, Williams CK, Newell KL, Ghetti B, DeTure MA, Dickson DW, Vinters HV, Felgner PL, Nakajima R, Glabe CG, Eisenberg DS. Crystal structure of a conformational antibody that binds tau oligomers and inhibits pathological seeding by extracts from donors with Alzheimer's disease. J Biol Chem. 2020 Jul 31;295(31):10662-10676. Epub 2020 Jun 3 PubMed.
- Silva MC, Nandi GA, Tentarelli S, Gurrell IK, Jamier T, Lucente D, Dickerson BC, Brown DG, Brandon NJ, Haggarty SJ. Prolonged tau clearance and stress vulnerability rescue by pharmacological activation of autophagy in tauopathy neurons. Nat Commun. 2020 Jun 26;11(1):3258. PubMed.
- Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, Neff N, Li SH, Rodriguez RD, Suemoto CK, Leite RE, Ehrenberg AJ, Pasqualucci CA, Seeley WW, Spina S, Heinsen H, Grinberg LT, Kampmann M. Molecular characterization of selectively vulnerable neurons in Alzheimer's disease. Nat Neurosci. 2021 Feb;24(2):276-287. Epub 2021 Jan 11 PubMed.
- Komatsu M, Chiba T, Tatsumi K, Iemura S, Tanida I, Okazaki N, Ueno T, Kominami E, Natsume T, Tanaka K. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 2004 May 5;23(9):1977-86. Epub 2004 Apr 8 PubMed.
- Wang L, Xu Y, Rogers H, Saidi L, Noguchi CT, Li H, Yewdell JW, Guydosh NR, Ye Y. UFMylation of RPL26 links translocation-associated quality control to endoplasmic reticulum protein homeostasis. Cell Res. 2020 Jan;30(1):5-20. Epub 2019 Oct 8 PubMed.
- Liang JR, Lingeman E, Luong T, Ahmed S, Muhar M, Nguyen T, Olzmann JA, Corn JE. A Genome-wide ER-phagy Screen Highlights Key Roles of Mitochondrial Metabolism and ER-Resident UFMylation. Cell. 2020 Mar 19;180(6):1160-1177.e20. Epub 2020 Mar 10 PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Samelson AJ, Ariqat N, McKetney J, Rohanitazangi G, ParraBravo C, Goodness D, Tian R, Grosjean P, Abskharon R, Eisenberg D, Kanaan NM, Gan L, Condello C, Swaney DL, Kampmann M. CRISPR screens in iPSC-derived neurons reveal principles of tau proteostasis. 2023 Jun 17 10.1101/2023.06.16.545386 (version 1) bioRxiv.
- ParraBravo C, Giani AM, Madero-Perez J, Zhao Z, Samelson AJ, Wong MY, Evangelisti A, Fan L, Pozner T, Mercedes M, Ye P, Patel T, Yarahmady A, Carling G, Lee VM, Sharma M, Mok S-A, Luo W, Zhao M, Kampmann M, Gong S, Gan L. Human iPSC 4R tauopathy model uncovers modifiers of tau propagation. 2023 Jun 22 10.1101/2023.06.19.544278 (version 1) bioRxiv.
Follow-On Reading
Papers
- Samelson AJ, Ariqat N, McKetney J, Rohanitazangi G, ParraBravo C, Bose R, Travaglini KJ, Lam VL, Goodness D, Dixon G, Marzette E, Jin J, Tian R, Tse E, Abskharon R, Pan H, Carroll EC, Lawrence RE, Gestwicki JE, Eisenberg D, Kanaan NM, Southworth DR, Gross JD, Gan L, Swaney DL, Kampmann M. CRISPR screens in iPSC-derived neurons reveal principles of tau proteostasis. 2024 Nov 04 10.1101/2023.06.16.545386 (version 4) bioRxiv.
Annotate
To make an annotation you must Login or Register.
Comments
University of Texas, Southwestern Medical Center
I think this is an interesting paper. It shows the power of the screening method to pick out genes with even subtle effects on cell biology. I would need a little more data to convince me that they have truly found modifiers of tau oligomer formation/clearance.
The reliance on conformation-specific antibodies should be supplemented with biochemistry to show that the putative oligomers detected are, in fact, higher-order tau assemblies.
The biological effects are quite subtle. Even in highly controlled conditions, the knockdown of Cul5 produces a ~2x increase in signal. This is surprisingly low if this is an important regulator. It would have been helpful to compare this to knockdown/inhibition of other parts of the degradation machinery for reference. Again, the use of biochemistry to study oligomer levels would have been useful to be more convincing.
The secondary point of the paper, that oxidative stress produces tau fragments, is interesting, but the meaning is unclear to me. Without carefully comparing the identity of the fragments created by stress to those that are detected as biomarkers in patients (e.g., by using mass spectrometry) it might be a stretch to suggest that oxidative stress is the cause of the biomarkers.
View all comments by Marc DiamondMake a Comment
To make a comment you must login or register.