Do APOE4’s Lipid Shenanigans Trigger Tauopathy?
Quick Links
Lipids are the hot new thing in Alzheimer’s research, with mounting evidence linking their dysregulation to amyloidosis, especially in the presence of the APOE4 allele. Now, researchers led by David Holtzman at Washington University in St. Louis and Gilbert Di Paolo at Denali Therapeutics, South San Francisco, tie lipids to tau pathology as well. In the November 22 Neuron, they report that cholesterol esters built up in glial cells of tauopathy mice expressing human APOE4. Getting lipids to leave these cells with the help of a liver-X receptor agonist dampened neuroinflammation, tau phosphorylation, and neurodegeneration.
"This work shows that APOE4 drives lipid accumulation in glia with strong downstream effects on inflammation and tau pathology. Importantly, this mechanism is independent of Aβ. It highlights a huge potential for next-generation LXR agonists as disease-modifying agents with fewer metabolic effects," commented Karen Gylys at University of California, San Francisco.
- In tauopathy mice, APOE4 causes cholesterol esters to build up in microglia.
- A drug that boosts lipid efflux lowered inflammation, p-tau, and neurodegeneration.
- Alas, it damages the liver.
Rik van der Kant at Vrije University, Amsterdam, agreed that these data point toward therapeutic avenues. “This paper will likely serve as a blueprint for future work aimed to mitigate ApoE4- and Tau-mediated lipid dysfunction … and is therefore in my opinion a major advance in the field,” he wrote to Alzforum (comment below).
Holtzman’s group had previously reported that APOE4 worsens tauopathy in mice by driving neuroinflammation (Sep 2017 news; Oct 2019 news; Apr 2021 news). Meanwhile, work from multiple groups had shown that APOE4 perturbs lipid metabolism in glial cells, spurring inflammation and amyloidosis (Apr 2019 conference news; Nov 2021 conference news).
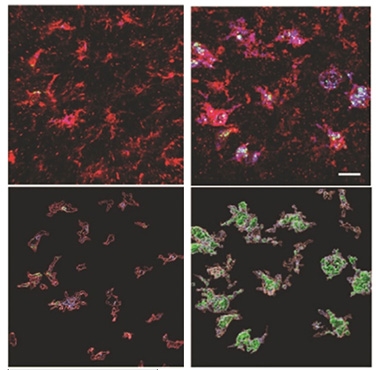
Fatty Buildup. In microglia (red), lysosomes (purple) fill up with cholesterol esters (green) in tauopathy mice expressing APOE4 (right), but not APOE3 (left). Bottom panels show 3D view. [Courtesy of Litvinchuk et al., Neuron.]
Holtzman wondered if faulty lipid metabolism in APOE4 carriers contributed to tauopathy, too. In collaboration with Denali researchers, first author Alexandra Litvinchuk profiled the lipid composition in the forebrains of P301S tau mice carrying human APOE3 or APOE4. At 9.5 months of age, the tauopathy E4 mice (TE4) had accumulated more cholesterol esters and phosphatidylcholine, an inflammatory lipid, than had their TE3 counterparts. Cholesterol esters filled microglial lysosomes (see image above). Oxidized phosphatidylcholines showed up in activated microglia and astrocytes. The amount of lipid buildup correlated with brain atrophy.
Could flushing out these lipids help the brain? To find out, Litvinchuk and colleagues fed TE4 mice the LXR agonist GW3965 starting at 6 months of age. This drug boosts expression of the lipid transporter ABCA1, which helps glia expel excess lipids. At 9.5 months, treated mice fared better than their untreated peers on multiple measures.
They had less inflammation, with microglial activation down by half, and astrocyte reactivity by a third. Fewer T cells invaded the hippocampus. Degeneration was slowed, with treated mice having about half as much phosphorylated tau, a third less plasma NfL, and a quarter less hippocampal atrophy (see image above). In addition, the LXR agonist prevented synapse loss and preserved the mice’s ability to build nests.

Clean Sweep. The hippocampus (top) and cortex (bottom) of TE4 mice (left) accumulate p-tau (black), but not when treated with an LXR agonist to promote lipid efflux (right). [Courtesy of Litvinchuk et al., Neuron.]
Gene expression studies confirmed that GW3965 treatment normalized glial lipid metabolism, as well as helping neurons maintain synapse function and better degrade proteins. Crossing TE4 mice with transgenics that expressed twice the normal amount of ABCA1 was similarly protective, showing that the treatment acted through this pathway.
The findings may translate to people, the authors noted. Di Paolo previously reported that Alzheimer’s disease brain has similar lipid abnormalities, accumulating cholesterol esters (Chan et al., 2012). Moreover, van der Kant, working with Lawrence Goldstein at the University of California, San Diego, found that cholesterol esters ramped up tau phosphorylation in induced neurons generated from people with AD (Feb 2019 news).
However, LXR agonists themselves are not a viable therapy. They trigger fatty acid buildup in the liver, van der Kant noted. “This paper solidifies the link between ApoE, cholesterol homeostasis and tau, but also highlights the need for more specific brain-cholesterol targeting interventions,” he wrote.—Madolyn Bowman Rogers
References
Mutations Citations
News Citations
- ApoE4 Makes All Things Tau Worse, From Beginning to End
- In Tauopathy, ApoE Destroys Neurons Via Microglia
- Squelching ApoE in Astrocytes of Tau-Ravaged Mice Dampens Degeneration
- Could Greasing the Wheels of Lipid Processing Treat Alzheimer’s?
- Do Lipids Lubricate ApoE's Part in Alzheimer Mechanisms?
- Cholesteryl Esters Hobble Proteasomes, Increase p-Tau
Research Models Citations
Paper Citations
- Chan RB, Oliveira TG, Cortes EP, Honig LS, Duff KE, Small SA, Wenk MR, Shui G, Di Paolo G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J Biol Chem. 2012 Jan 20;287(4):2678-88. PubMed.
Further Reading
News
- Taming ApoE Via the LDL Receptor Calms Microglia, Slows Degeneration
- Can Flipping a Lipid Switch Protect the Brain?
- Cracking the Cholesterol-AD Code: Metabolites and Cell Type
- Lipid-Laden, Sluggish Microglia? Blame Aβ.
- Newly Identified Microglia Contain Lipid Droplets, Harm Brain
- ApoE4 Glia Bungle Lipid Processing, Mess with the Matrisome
- Sans TREM2, ApoE4 Drives Microgliosis and Atrophy in Tauopathy Model
Primary Papers
- Litvinchuk A, Suh JH, Guo JL, Lin K, Davis SS, Bien-Ly N, Tycksen E, Tabor GT, Remolina Serrano J, Manis M, Bao X, Lee C, Bosch M, Perez EJ, Yuede CM, Cashikar AG, Ulrich JD, Di Paolo G, Holtzman DM. Amelioration of Tau and ApoE4-linked glial lipid accumulation and neurodegeneration with an LXR agonist. Neuron. 2024 Feb 7;112(3):384-403.e8. Epub 2023 Nov 22 PubMed. Neuron.
Annotate
To make an annotation you must Login or Register.
Comments
Vrije Universiteit Amsterdam
The 2017 Nature paper by the Holtzman group describing their findings that ApoE accelerates Tau pathology (Shi et al., 2017), even in the absence of amyloid pathology (van der Kant et al., 2020), has been a major conceptual breakthrough in understanding the role of ApoE in Alzheimer’s pathology. With impressive follow-up through a number of papers, the Holtzman lab and others have probed which cell types are involved, and which other factors—e.g., diet, sleep—might modify the ApoE4 effect on Tau.
I’m excited by this new study because it dives deeper into the mechanisms of how ApoE4 might affect Tau. A very plausible candidate for these effects is altered lipid/cholesterol metabolism. ApoE4 is the main brain cholesterol carrier, and is known to disrupt cholesterol metabolism in different model systems and cell types (Blanchard et al., 2022; TCW et al., 2019). Furthermore, we showed that cholesterol accumulation in neurons drives Tau accumulation (van der Kant et al., 2019), while more recently Tau mutations have been shown to regulate cholesterol homeostasis (Glasauer et al., 2022; Szabo et al., 2023).
Interestingly, the authors report here for the first time that cholesterol metabolism is indeed significantly altered in P301S/ApoE4 transgenic mice. Through a series of well-crafted lipidomic experiments, they show changes in cholesterol, its storage products, but also changes in lysosomal lipids. More importantly, they show rescue of many of these phenotypes by an LXR agonist and ABCA1 overexpression, both known to drive cholesterol export. The authors go on to show that the LXR agonist can partially rescue Tau pathology and inflammation in these mice, as well as improve behavioral deficits.
This paper provides an important proof of concept for the role cholesterol has in mediating the effects of ApoE4 on Tau pathology. This effect might be driven through glial lipid changes and downstream inflammatory processes, as recent work has uncovered the importance of lipids in glial activation. The authors also report less phosphorylated Tau in the neurons (and more proteasome), which could also indicate neuronal cholesterol-dependent effects on the proteasome and pTau degradation, as we have shown in iPSC-neurons (van der Kant et al., 2019).
LXR agonists have been clinically explored for the treatment of atherosclerosis, but have not reached the market due to severe off-target effects on fatty acid metabolism, i.e., fatty liver phenotypes. Indeed, the authors also see increased fatty acid accumulation in the brains of the LXR-treated P301S/ApoE4 mice. This paper therefore solidifies the link between ApoE, cholesterol homeostasis and Tau, but also highlights the need for more specific brain-cholesterol targeting interventions. This paper will likely serve as a blueprint for future work aimed to mitigate ApoE4- and Tau-mediated (lipid) dysfunction by candidate interventions
References:
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C, Alzheimer’s Disease Neuroimaging Initiative, Fagan AM, Miller BL, Boxer AL, Seeley WW, Butovsky O, Barres BA, Paul SM, Holtzman DM. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017 Sep 28;549(7673):523-527. Epub 2017 Sep 20 PubMed.
van der Kant R, Goldstein LS, Ossenkoppele R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020 Jan;21(1):21-35. Epub 2019 Nov 28 PubMed.
Blanchard JW, Akay LA, Davila-Velderrain J, von Maydell D, Mathys H, Davidson SM, Effenberger A, Chen CY, Maner-Smith K, Hajjar I, Ortlund EA, Bula M, Agbas E, Ng A, Jiang X, Kahn M, Blanco-Duque C, Lavoie N, Liu L, Reyes R, Lin YT, Ko T, R'Bibo L, Ralvenius WT, Bennett DA, Cam HP, Kellis M, Tsai LH. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature. 2022 Nov;611(7937):769-779. Epub 2022 Nov 16 PubMed. Correction.
TCW J, Liang SA, Qian L, Pipalia NH, Chao MJ, Shi Y, Bertelsen SE, Kapoor M, Marcora E, Sikora E, Holtzman DM, Maxfield FR, Zhang B, Wang M, Poon WW, Goate AM. Cholesterol and matrisome pathways dysregulated in human APOE ε4 glia. 2019 Jul 25 10.1101/713362 (version 1) bioRxiv.
van der Kant R, Langness VF, Herrera CM, Williams DA, Fong LK, Leestemaker Y, Steenvoorden E, Rynearson KD, Brouwers JF, Helms JB, Ovaa H, Giera M, Wagner SL, Bang AG, Goldstein LS. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-β in iPSC-Derived Alzheimer's Disease Neurons. Cell Stem Cell. 2019 Mar 7;24(3):363-375.e9. Epub 2019 Jan 24 PubMed.
Glasauer SM, Goderie SK, Rauch JN, Guzman E, Audouard M, Bertucci T, Joy S, Rommelfanger E, Luna G, Keane-Rivera E, Lotz S, Borden S, Armando AM, Quehenberger O, Temple S, Kosik KS. Human tau mutations in cerebral organoids induce a progressive dyshomeostasis of cholesterol. Stem Cell Reports. 2022 Sep 13;17(9):2127-2140. Epub 2022 Aug 18 PubMed.
Szabo L, Cummins N, Paganetti P, Odermatt A, Papassotiropoulos A, Karch C, Götz J, Eckert A, Grimm A. ER-mitochondria contacts and cholesterol metabolism are disrupted by disease-associated tau protein. EMBO Rep. 2023 Aug 3;24(8):e57499. Epub 2023 Jul 4 PubMed.
University of California, San Francisco
In this paper, the Holtzman group continues to systematically dissect ApoE4 involvement in tau-mediated neurodegeneration and inflammation. This set of experiments highlights the critical role of lipid metabolism in the progression of pathology and builds on other recent work showing that a toxic accumulation of microglial cholesterol esters is a central feature of the lipid mechanism.
An important missing link in the mechanism is what’s happening in neurons and when. There is no final consensus on LXR expression in adult neurons, but considering neuronal cell-cycle re-entry in AD, what would be the direct LXR agonist effect on neuronal cells? Work from our lab shows a significant reduction in cholesterol esters in surviving synapses from the human AD cortex (Bilousova et al., 2019) and earlier work showed accumulations of both free cholesterol and ganglioside GM1 in synapses positive for Aβ (Gylys et al., 2007). These synaptic changes seem likely to be downstream from lipid changes in microglia and astrocytes and may not be related. But these changes in synapses, which are enveloped by astrocytic and microglial processes, illustrate the complexity of understanding APOE-mediated lipid changes in disease progression.
The Litvinchuk paper also points to the potential of LXR agonists as disease-modifying therapies and the urgent need for next-generation LXR agonists without the peripheral effects of GW3965 and T0901317 on cholesterol metabolism, triglycerides, and insulin secretion by pancreatic beta-cells (Maczewsky et al., 2020).
—Tina Bilousova, University of California, San Francisco, is the co-author of this comment.
References:
Bilousova T, Melnik M, Miyoshi E, Gonzalez BL, Poon WW, Vinters HV, Miller CA, Corrada MM, Kawas C, Hatami A, Albay R 3rd, Glabe C, Gylys KH. Apolipoprotein E/Amyloid-β Complex Accumulates in Alzheimer Disease Cortical Synapses via Apolipoprotein E Receptors and Is Enhanced by APOE4. Am J Pathol. 2019 Aug;189(8):1621-1636. Epub 2019 May 17 PubMed.
Gylys KH, Fein JA, Yang F, Miller CA, Cole GM. Increased cholesterol in Abeta-positive nerve terminals from Alzheimer's disease cortex. Neurobiol Aging. 2007 Jan;28(1):8-17. PubMed.
Maczewsky J, Kaiser J, Krippeit-Drews P, Drews G. Approved LXR agonists exert unspecific effects on pancreatic β-cell function. Endocrine. 2020 Jun;68(3):526-535. Epub 2020 Mar 7 PubMed.
University of Pittsburgh
University of Pittsburgh
This article presents the therapeutic effects of the LXR ligand GW3965 in 9.5-month-old tau transgenic mice expressing human E3 (TE3) or E4 (TE4) as well as their E3 and E4 non-transgenic controls. The authors evaluated several parameters indicative of neurodegeneration: 1) nest-building behavior (generalized as behavioral deficit); 2) p-tau levels, without affecting APOE levels; 3) glial reactivity, T cell infiltration, and inflammation, and 4) synaptic loss. They also report that the application of GW3965 changed the transcriptional response in TE4 mice. Additionally, they performed lipidomics on forebrain tissue and on isolated microglia and astrocytes. In forebrain, they found that several species of cholesteryl esters were increased in TE4 versus TE3 mice. Interestingly, the same species were decreased in E4 versus E3 mice, suggesting that tau pathology may contribute to this effect. In contrast, cholesterol sulfate, and several phospholipids and diacylglyceride species, were decreased in TE4 versus TE3 mice.
Then, based on what the authors found after application of a pharmacologically active synthetic ligand of nuclear receptors LXR/RXR, they conclude that “promoting efflux of glial lipids may serve as a therapeutic approach to ameliorate tau and ApoE4-linked neurodegeneration.”
There have been dozens of studies demonstrating that promoting the efflux of major brain cells' lipids should be considered a rational and physiologically justifiable therapeutic approach in Alzheimer's disease. The mere fact that a synthetic LXR ligand, as the authors have demonstrated, may have a therapeutic effect in a model of tauopathy is a significant contribution to the understanding of the role of LXR-responsive genes in neurodegeneration, and reopens the door for exploring therapeutic strategies proposed years ago.
The list of remarkable responses to an experimental therapeutic agent in a mouse model of tauopathy is probably unique among many disorders, not only neurodegenerative ones. In a complex mouse model expressing mutant tau and human APOE4, a reasonable explanation of the plethora of ligand-activated LXR/RXR effects is through the LXR/RXR-ABCA1-APOE regulatory axis. While the methods used here did not reveal upregulation of APOE—neither at genomic nor at protein level—clearly a significant part of the effects of the synthetic ligand is mediated by ABCA1 upregulation.
Importantly, the effects of GW3965 mimic the phenotype of ABCA1 transgenic mice. Considering the limited, but extremely important functions of ABCA1 in brain, the results of this study will doubtlessly stimulate a new wave of research on the decades-old and valid hypothesis that major aspects of brain lipid metabolism depend on the LXR/RXR-ABCA1 regulatory axis with significant impact on neurodegeneration.
University of British Columbia
There is a complex relationship between lipid metabolism and AD pathophysiology. ApoE is a major genetic risk factor for late-onset AD and the major lipid carrier in the central nervous system. In the brain, apoE also mediates the removal of cholesterol in a process known as cholesterol efflux. This is particularly important for phagocytic cells such as microglia that ingest vast amounts of lipids, and impaired or saturated cholesterol efflux can trigger other cellular responses including altered lysosomal function and transcriptomic profiles. On the other hand, augmenting cholesterol efflux capacity, which can be accomplished by genetic or pharmacological means, is generally considered beneficial.
The best-studied class of molecules that promote expression of genes involved in cholesterol efflux are agonists of the liver X receptor (LXR), which stimulate expression genes such as ABC transporter cassette A1 (ABCA1) and apolipoprotein cholesterol acceptors such as apoE and apoA1 (Hua and Wei, 2023). Previous studies evaluating LXR agonists in AD animal models focused mainly on amyloid pathways (Donkin et al., 2010; Fitz et al., 2014; Lefterov et al., 2007; Terwel et al., 2011).
Here, Alexandra Litvinchuk from David Holtzman’s laboratory investigated the effect of the LXR agonist GW3965 as well as overexpression of ABCA1 on tau and apoE4-linked glial lipid accumulation and neurodegeneration. Using targeted lipidomics, they found that 9.5-month-old P301S tau mice expressing human apoE4 had elevated cholesterol esters in the forebrain, increased lipid accumulation in microglia, and altered cholesterol metabolism in microglia and astrocytes. Initiation of oral GW3965 treatment at 6 months of age attenuated neurodegeneration, improved nesting behavior, and reduced tauopathy as measured by AT8 and ptau immunostaining. As expected from previous reports, there was no change in brain apoE levels, suggesting that changes in apoE lipidation status rather than apoE expression were driving the effects observed.
Intriguingly, GW3965 treatment also reduced T-cell infiltration and inflammation. This observation is of key importance as peripheral lipoprotein-mediated signaling may be involved in regulating T-cell infiltration to the CNS and systemic administration of GW3965 would also affect peripheral pathways. Litvinchuk also found that selective overexpression of ABCA1 in the brain also reduced neurodegeneration, tauopathy, microglial activation, and lipid accumulation in tau-apoE mice, suggesting that enhancing cholesterol efflux selectively in the brain is sufficient to drive many of the beneficial effects observed. However, T-cell infiltration was not reported for this experiment. It will be interesting to further explore this component, as peripheral lipoprotein subclasses have complex relationships with inflammatory and immunomodulatory pathways including Tregs (Pinzon Grimaldos et al., 2022) and oral GW3965 administration would affect peripheral cholesterol efflux but brain-specific ABCA1 overexpression would not.
One challenge for future translation into clinical utility is that small-molecule LXR agonists have several adverse effects in humans, including stimulation of hepatic lipogenesis that increased pro-atherogenic low-density-lipoprotein levels. One potential solution could be administration directly to the CNS to limit systemic exposure. Another solution could involve capitalizing on the synergy between circulating high density lipoproteins and LXR agonism (Morin et al., 2020), if further research supports a role for peripheral lipoproteins on immune cell access to the CNS and tau-mediated neurodegeneration.
Make a Comment
To make a comment you must login or register.