Doubling Rab5 in Mice Leads to Neurodegeneration—Without Plaques
Quick Links
Neurons crowded with distended endosomes are a hallmark of Alzheimer’s disease, but does this malfunction drive the neurodegenerative cascade? A new transgenic mouse, described last November 24 in Cell Reports, supports the possibility. Researchers led by Ralph Nixon at New York University describe how overexpression of Rab5 in neurons caused endosomes to swell and build up. It also caused other AD-like features as the mice aged, including synaptic deficits, tau hyperphosphorylation, and withering cholinergic neurons. This chain of events unfolded in the absence of Aβ accumulation, suggesting the endosomal malfunctions were sufficient to bring on key characteristics of AD.
- Expressing human Rab5 in neurons causes endosomes to swell.
- The transgenic mice have synaptic deficits and hyperphosphorylated tau.
- Cholinergic neurons and fibers degenerate; memories fade.
“This paper provides additional support to implicate endosomal trafficking in AD, and shows this pathway is a convergence point for both APP-dependent and APP-independent mechanisms of disease,” commented Scott Small of Columbia University in New York (full comment below).
“The studies … suggest that endosomal dysfunction is a contributor to both the prodromal and the neurodegenerative phases of AD, the latter of which has commonly been attributed to the consequences of one or more of extracellular Aβ accumulation, amyloid plaque deposition, and tau aggregation,” wrote Rick Livesey and Christy Hung of University College London. “Their findings also open up new avenues to explore whether Rab5 overactivation and its signaling effectors might be potential targets for therapeutic interventions against AD or other neurodegenerative conditions involving endosomal-lysosomal dysfunction.” (Full comment below.)
Endosomal abnormalities emerge early in Alzheimer’s, prior to overt accumulation of Aβ (Cataldo et al., 2000). The endosomal swellings have been tied to the β-CTF fragment of amyloid precursor protein (APP). The product of BACE1 cleavage, this fragment has been implicated in the recruitment of Rab5 to endosomes, where the small GTPase kicks on and fuels endocytosis (Jul 2015 news). The β-CTF-Rab5 pathway has been blamed for endosomal dysfunction in cell culture models of familial AD, as well as in a mouse model of Down’s syndrome, which expresses an extra copy of APP (Aug 2019 news; Jiang et al., 2019; Alam et al., 2017).
Might endosomal malfunctions drive neurodegeneration, even when Aβ is not accumulating? To address this question, first author Anna Pensalfini and colleagues generated a mouse that overexpresses a Myc-tagged version of human Rab5 in neurons. The mice overexpressed Rab5 by about 2.5-fold compared to wild-type mice, on par with the elevated levels of the GTPase in the AD brain (Ginsberg et al., 2010). Using multiple approaches, the researchers reported that this overexpression of Rab5 also led to its activation—the transgenic mice had twice the amount of GTP-bound Rab5 lodged in their neuronal endosomes as did their wild-type counterparts.
As the authors describe it, this pathologically active (PA) Rab5 ratcheted up endocytosis in the hippocampus and cortex. In 6- to 8-month-old PA-Rab5 mice, swollen endosomes took up twice as much space within neurons as they did in neurons from wild-type. The endosomes crowded neuronal cell bodies and dendrites, resembling the endosomal pathology seen in the human Alzheimer’s brain. The endosomal dysfunction did not significantly affect levels of APP metabolites—including β-CTF and Aβ peptides.
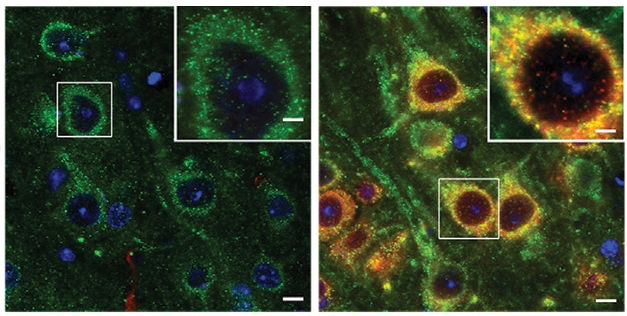
Endosome Invasion. Endosomes in human Rab5-expressing neurons (red) took up twice as much space as those in wild-type neurons (left) with only mouse Rab5 (green). [Courtesy of Pensalfini et al., Cell Reports, 2020.]
Besides endosomal distension, what else happened? The researchers found that the excess Rab5 triggered synaptic snafus. In hippocampal brain slices from 6-month-old mice, both long-term potentiation and depression—the yin and yang of synaptic sensitivity that makes synapses so plastic—were derailed. The researchers found fewer AMPA receptors on neuronal cell membranes, probably because the revved-up endocytosis snatched them from the surface. The investigators detected all manner of defects in dendritic spines. Depending on the region in question, spines were either fewer or shorter—morphological changes that have been observed in AD mouse models and in the human Alzheimer’s brain.
Rab5 overexpression messed with AKT, a kinase required for pro-survival signaling. In so doing, Rab5 switched on GSK-3β, a kinase downstream of AKT that is known to phosphorylate tau. And sure enough, the PA-Rab5 mice accumulated hyperphosphorylated tau in different regions of the cortex. As measured by immunofluorescence using two different tau antibodies, PHF1S396/404 and CP13S202, the researchers spotted Rab5+ swollen endosomes co-localized with phospho-tau within neurons.
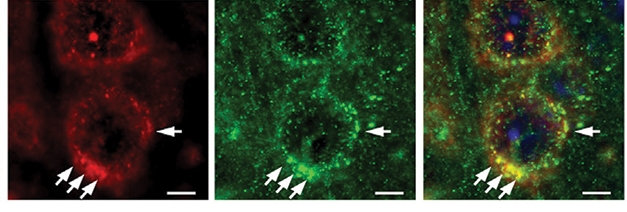
Tussles without Tangles. In human Rab5-expressing mice, phospho-tau (red) accumulates in cortical neuron endosomes (left), where it tussles with Rab5 (green, center). Right panel shows both. [Courtesy of Pensalfini et al., 2020.]
Did Rab5 lead to neurodegeneration? In the PA-Rab5 mice, the researchers detected a gradual loss of basal forebrain cholinergic neurons as gauged by staining for choline acetyltransferase (ChAT), starting at 7 months of age. They monitored these neurons because they are the first to die in people with AD, which could be because they rely on endosome-mediated, retrograde transport of growth factors made in the hippocampus. Cholinergic fibers withered, and both their cell bodies and dendrites choked up with swollen endosomes. All the while, endosomal dysfunction and other signs of cellular distress, including autophagic structures crammed into dendrites and synaptic terminals, manifested in hippocampal regions that receive cholinergic input. Together, the findings suggest that the endosomal dysfunction triggered by Rab5 overexpression dismantled key cholinergic circuitry.
The PA-Rab5 mice also showed signs of memory loss. At 6 months, compared to wild-type, they were less adept at distinguishing between novel and familiar objects.
Overall, the findings suggest that Rab5 overactivation leads to several characteristics of AD, including synaptic dysfunction, tau hyperphosphorylation, and degeneration of the cholinergic system, Nixon said.
“Although numerous in-vitro experiments have indirectly linked hyperactivation of Rab5 to cellular pathology in AD, this is the first in-vivo study that has established a causal effect of Rab5-endosomal dysfunction on AD-like cellular pathology, and synaptic and behavioral deficits in mice,” wrote Chengbiao Wu of the University of California, San Diego. “This study is timely and consistent with the fact that many newly identified AD risk factors are involved in regulating endocytic trafficking.”
Nixon stressed that in the context of AD, β-CTF and likely a complex mix of other factors are responsible for stoking Rab5. William Mobley of the University of California, San Diego, called the quality of the work impressive. He also noted that this transgenic model does not recapitulate what happens in AD or DS, where Rab5 is disproportionately activated while its expression remains normal. Mobley thinks it’s unclear how Rab5 becomes activated in disease.
Mobley is investigating if Aβ itself triggers an uptick in β-CTF, setting off a feedback loop that promotes Rab5 overactivation and endosomal dysfunction. In the PA-Rab5 mouse, Rab5 is constitutively overactivated, rendering the model unable to address what activates the GTPase in AD. Mobley also wondered how Rab5 overexpression affects other parts of the endolysosomal system, noting that defects in lysosomal digestion, which are also known to occur in AD and DS, may also damage neurons.
For other scientists’ perspective on the data, see comments below.
The findings do not rule out that Aβ contributes to endolysosomal dysfunction, Nixon stressed, noting that later parts of the pathway, including lysosomal digestion, likely play a role in disease. He thinks Rab5 makes a promising therapeutic target for AD and related diseases. A recent trial of neflamapimod, an inhibitor of the p38 MAP kinase that overactivates Rab5, reported a benefit in dementia with Lewy bodies, and is still on the table as an investigational AD therapeutic. That said, Nixon believes BACE inhibitors, which reduce levels of β-CTF as well as Aβ, remain an obvious choice. Though these inhibitors had side effects in multiple clinical trials, lower doses would suffice to reduce β-CTF enough to halt downstream endosomal dysfunction (Dec 2020 news).—Jessica Shugart
References
News Citations
- Partners in Crime: APP Fragment and Endosomal Protein Impair Endocytosis
- Familial AD Mutations, β-CTF, Spell Trouble for Endosomes
- New Data from Past BACE Inhibitor Trials Shed Light on Side Effects
Research Models Citations
Therapeutics Citations
Paper Citations
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000 Jul;157(1):277-86. PubMed.
- Jiang Y, Sato Y, Im E, Berg M, Bordi M, Darji S, Kumar A, Mohan PS, Bandyopadhyay U, Diaz A, Cuervo AM, Nixon RA. Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-βCTF (C99). J Neurosci. 2019 Jul 3;39(27):5255-5268. Epub 2019 May 1 PubMed.
- Alam J, Jiang Y, Nixon R. Antagonism of p38 MAPK Alpha (p38α) Reverses APP-Induced Endosomal Abnormalities and Improves Lysosomal Function in Down Syndrome Fibroblasts. Alzheimer's & Dementia Supplement, July 2017
- Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y, Wuu J, Chao MV, Mufson EJ, Nixon RA, Che S. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer's disease progression. Biol Psychiatry. 2010 Nov 15;68(10):885-93. Epub 2010 Jul 23 PubMed.
Further Reading
Papers
- Germann UA, Alam JJ. P38α MAPK Signaling-A Robust Therapeutic Target for Rab5-Mediated Neurodegenerative Disease. Int J Mol Sci. 2020 Jul 31;21(15) PubMed.
Primary Papers
- Pensalfini A, Kim S, Subbanna S, Bleiwas C, Goulbourne CN, Stavrides PH, Jiang Y, Lee JH, Darji S, Pawlik M, Huo C, Peddy J, Berg MJ, Smiley JF, Basavarajappa BS, Nixon RA. Endosomal Dysfunction Induced by Directly Overactivating Rab5 Recapitulates Prodromal and Neurodegenerative Features of Alzheimer's Disease. Cell Rep. 2020 Nov 24;33(8):108420. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University College London
This paper from the Nixon group is a thought-provoking addition to a growing body of evidence that dysfunction of the endolysosomal system in neurons is an early, causal, pathogenic process in Alzheimer’s disease, and not a secondary consequence of protein aggregation. Much of that pioneering work has come from the Nixon group, beginning with seminal findings of endosome enlargement in forebrain neurons in individuals with dementia due to APP duplications or trisomy 21/Down syndrome.
Previously, it was established that APP processing by gamma-secretase to generate the C-terminal fragment of APP (beta-CTF) is central to endosome enlargement and dysfunction in Alzheimer’s disease and Down syndrome. However, the process by which it does so was somewhat unclear, although work from the Nixon group and Chengbiao Wu at UCSD indicated that increased beta-CTF-mediated endolysosomal dysfunction was dependent on Rab5. The new work uses transgenic mouse models, over-expressing Rab5a in neurons, to demonstrate that this leads to increased activation of Rab5a, which in turn causes neuronal endolysosome dysfunction, independent of any changes in APP processing. This firmly places Rab5a activation downstream of beta-CTF in the pathogenic pathway, and is very well characterized by the authors, combining biochemistry and cell biology.
A strength of the paper is that the authors go on to use the new mouse model to explore different pathological consequences for increased Rab5a activation and endosome dysfunction on neuronal physiology, including glutamate receptor recycling and synaptic plasticity, GSK3-beta signaling, and tau phosphorylation. Those studies demonstrate a plethora of changes in neuronal cell biology that occur downstream of Rab5a activation, and suggest that endosomal dysfunction is a possible contributor to both the prodromal and the neurodegenerative phases of AD, the latter of which commonly has been attributed to the consequences of one or more of extracellular Aβ accumulation, amyloid plaque deposition, and tau aggregation.
Their findings also open up new avenues to further explore whether Rab5 overactivation and its signaling effectors might be potential targets for therapeutic interventions against AD and in other neurodegenerative conditions involving endosomal-lysosomal dysfunction.
—Christy Hung is co-author of this comment.
Lund University
This is a very interesting article by Pensalfini and colleagues from the lab of an outstanding researcher in our field, Randy Nixon. Over several decades his group has pioneered the important role of the endosome-lysosome-autophagy system in Alzheimer’s pathogenesis.
In this excellent study, they show that a mouse overexpressing Rab5 recapitulates numerous features of Alzheimer’s disease (AD), including enlarged early endosomes, alterations in tau, synapses, spines, and in synaptic plasticity, memory deficits, and even neurodegeneration of cholinergic neurons.
The concept that endocytic alterations can promote AD is strengthened by this work, and by genetics studies implicating the endocytic system in AD. As with all good papers, one does have questions; in particular, one wonders by what mechanism(s) Rab5 becomes overactive with aging to promote human AD and also how other neurons fare that are known to be vulnerable to early damage in AD, such as entorhinal layer 2 neurons.
In landmark papers Nixon and colleagues years ago described early, pre-plaque enlargement of Rab5 endosomes with AD pathogenesis. Nixon’s and other groups have stressed the involvement of APP β-C-terminal fragments in inducing this endosome enlargement. Aβ’s involvement in endosome changes was often excluded by lack of protection against such changes by γ-secretase inhibition, which blocks production of Aβ but not of APP CTFs. However, this is a complex manipulation affecting numerous substrates, and given the potential toxicity of such inhibition it does not really exclude a role for Aβ. Moreover, we and others have shown that Aβ itself can directly promote endosome enlargement and that altering endosome biology can promote AD-like Aβ changes.
In the current study the authors indicate that in their Rab5-overexpressing mice, endosome changes are independent of APP/Aβ. However, looking at their bar graph in Supplemental Figure S1, there appear to be considerable trends for increased brain Aβ; particularly of Aβ42. Moreover, while the authors stress enlarged endosomes as the earliest pathobiological change seen in prodromal/preclinical AD, one wonders how this change relates temporally to the well-established and very early preclinical drop of Aβ42 in CSF.
University of California San Diego
This new study by Pensalfini and colleagues has elegantly demonstrated that a modest increase in Rab5 level via transgenic expression of a myc-tagged human Rab5a construct, driven by the neuronal Thy-1 promoter (PA-Rab5 mice), was sufficient to induce synaptic dysfunction and behavioral deficits in mice.
Although numerous in vitro experiments have indirectly linked hyperactivation of Rab5 to cellular pathology in AD, this is the first in-vivo study that has established a causal effect of Rab5-endosomal dysfunction on AD-like cellular pathology and synaptic and behavioral deficits in mice. This study is timely and consistent with the fact that many newly identified AD risk factors are involved in regulating endocytic trafficking.
Dysregulation of these risk factors represents an important area in our understanding of AD pathogenesis, especially in late-onset AD. For example, RIN3 (Ras And Rab Interactor 3) is a guanine nucleotide exchange factor (GEF) that activates members of the Rab5 family. RIN3 expression is likely increased in AD patients (Kunkle et al., 2017; Pathak et al., 2019). We have recently confirmed RIN3 expression was upregulated at the embryonic stages in the APP/PS1 mouse model of AD (Shen et al., 2020), which is consistent with previous studies demonstrating that Rab5 activation is a manifestation of early endocytic dysfunction in AD. We have further demonstrated that RIN3 formed a complex with two other AD risk factors, BIN1 and CD2AP; RIN3-BIN1 increased the level of tau-PHF1 while RIN3-CD2AP enhanced APP processing to yield APP-CTFs. Both pathways are mediated by hyperactivation of Rab5 since the effects were effectively blocked by the dominant and negative Rab5 S34N construct.
The finding that PA-Rab5 mice showed an increase in tau-PHF1 through GSK3β is very interesting; However, one surprising finding in the Pensalfini paper is that APP processing was not impacted in the PA-Rab5 mice. One would think that increased endocytosis will lead to more surface APP internalized and trapped in the endocytic pathways, which in turn would result in enhancement of APP cleavage by BACE.
It will be very interesting to find out why this is not the case in the PA-Rab5 mice.
References:
Kunkle BW, Vardarajan BN, Naj AC, Whitehead PL, Rolati S, Slifer S, Carney RM, Cuccaro ML, Vance JM, Gilbert JR, Wang LS, Farrer LA, Reitz C, Haines JL, Beecham GW, Martin ER, Schellenberg GD, Mayeux RP, Pericak-Vance MA. Early-Onset Alzheimer Disease and Candidate Risk Genes Involved in Endolysosomal Transport. JAMA Neurol. 2017 Sep 1;74(9):1113-1122. PubMed.
Pathak GA, Silzer TK, Sun J, Zhou Z, Daniel AA, Johnson L, O'Bryant S, Phillips NR, Barber RC. Genome-Wide Methylation of Mild Cognitive Impairment in Mexican Americans Highlights Genes Involved in Synaptic Transport, Alzheimer's Disease-Precursor Phenotypes, and Metabolic Morbidities. J Alzheimers Dis. 2019;72(3):733-749. PubMed.
Shen R, Zhao X, He L, Ding Y, Xu W, Lin S, Fang S, Yang W, Sung K, Spencer B, Rissman RA, Lei M, Ding J, Wu C. Upregulation of RIN3 induces endosomal dysfunction in Alzheimer's disease. Transl Neurodegener. 2020 Jun 18;9(1):26. PubMed.
Columbia University
This is a beautiful paper by Randy Nixon, whose group first described enlarged endosomes as a cytopathogenic feature of AD. To date, enlarged endosomes have been shown to occur via the genes that cause early onset AD in an APP dependent manner (e.g., Kwart et al., 2019); or by the one gene that appears to cause late-onset AD, the retromer-receptor Sorl1, in an APP independent manner (Knupp et al., 2020).
In this tour de force, the elegant series of studies show that overexpression of RAB5 can also cause enlarged endosomes in an APP-independent manner, and recapitulates many other features of AD.
This paper provides additional support that endosomal trafficking is a biological pathway implicated in AD, and that this pathway is a convergence point for both APP-dependent and APP-independent mechanisms of disease.
References:
Kwart D, Gregg A, Scheckel C, Murphy EA, Paquet D, Duffield M, Fak J, Olsen O, Darnell RB, Tessier-Lavigne M. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP β-CTFs, Not Aβ. Neuron. 2019 Oct 23;104(2):256-270.e5. Epub 2019 Aug 12 PubMed.
Knupp A, Mishra S, Martinez R, Braggin JE, Szabo M, Kinoshita C, Hailey DW, Small SA, Jayadev S, Young JE. Depletion of the AD Risk Gene SORL1 Selectively Impairs Neuronal Endosomal Traffic Independent of Amyloidogenic APP Processing. Cell Rep. 2020 Jun 2;31(9):107719. PubMed.
KULeuven & VIB
This new study in Cell Reports builds further on the contribution of Rab5 overactivation in relation to AD onset and progression. Thus far, Rab5 hyperactivation has been observed by several groups in vitro, including in human neurons differentiated from AD patient or gene-edited iPSCs, and triggered by the overaccumulation of APP β-CTF. However, an in vivo validation was lacking and in particular the contribution of Rab5 overactivation (OA) in the disease progression.
The authors generated a transgene model with moderate overexpression of Rab5a, mimicking the OA observed in other models including of Down’s syndrome (DS). The exciting aspect is that in this way they succeeded in disconnecting the contribution of APP b-CTFs from that of OA of Rab5.
Remarkably, a mere OA of Rab5a seems to recapitulate several features of not only prodromal and onset stages of AD pathology, including endosomal pathology, but of the neurodegenerative cascade as well. Although Rab5 is itself not popping up as a risk factor, this a potential interesting model to study the neurodegenerative phenotype in AD pathology independently from the amyloid/APP β-CTF burden, and maybe as relevant as, for instance, NPC1 knockout is for modelling the related lysosomal storage diseases.
However, and although Rab5 OA on its own induces characteristic enlarged endosomes that are observed in AD cytopathology, it cannot be excluded that in a true AD context, it is a bystander in the causality of this phenotype. Whereas Rab5 OA suggests a derailment in internalization and early endosomal sorting, a delay in cargo exit also explains endosome swelling. In this respect there is a remarkable resemblance to the effects of SorlA deficiency on endosomal swelling and trafficking defects (Knupp et al., 2020). Being related to retromer function, these findings link the observed endosomal phenotype more to cargo recycling defects. It would be interesting to explore the Rab5 OA model for these aspects of endosomal traffic jams, and by extension, lysosomal functions. Several groups including ours (Peric and Annaert, 2015), but also Scott Small and Gregory Petsko’s work, highlighted a more prominent role for cargo jamming and recycling defects that better explain the endosomal pathology observed in AD.
By overexpressing Rab5 in CNS neurons, the authors also highlight a possible more original neuronal contribution in prodromal and early stage of AD pathology. This is further corroborated by the SorLA KO study, where endosomal swellings were only observed in human neurons differentiated from SorlA KO iPSCs, but not microglia. This is an important aspect given the current shift to the contribution of microglia in AD pathoethiology. Also, other risk genes may affect similar steps in endosomal transport regulation (including lipid transfer between these organelles). Importantly, both this and the Knupp SorLA study also dissociate the endosomal defects from amyloidogenic APP processing: therefore, they support an emerging picture that sporadic AD-related endosomal transport dysregulation converges with early onset AD-related APP processing on the same neuronal AD pathology (Peric and Annaert, 2015).
Given that endosomal defects are among the earliest cytopathological signs in AD, likely occurring first in neurons, the cellular phase in AD (De Strooper and Karran, 2016) may be considered to initiate ahead of, or at most in parallel with, the biochemical phase of sporadic AD.
References:
Knupp A, Mishra S, Martinez R, Braggin JE, Szabo M, Kinoshita C, Hailey DW, Small SA, Jayadev S, Young JE. Depletion of the AD Risk Gene SORL1 Selectively Impairs Neuronal Endosomal Traffic Independent of Amyloidogenic APP Processing. Cell Rep. 2020 Jun 2;31(9):107719. PubMed.
Peric A, Annaert W. Early etiology of Alzheimer's disease: tipping the balance toward autophagy or endosomal dysfunction?. Acta Neuropathol. 2015 Mar;129(3):363-81. Epub 2015 Jan 3 PubMed.
De Strooper B, Karran E. The Cellular Phase of Alzheimer's Disease. Cell. 2016 Feb 11;164(4):603-15. PubMed.
Tel Aviv University
In this excellent study the Nixon group show that a mouse overexpressing Rab5 recapitulates numerous features of Alzheimer’s disease, including enlarged early endosomes, alterations in tau, synapses, spines, and in synaptic plasticity, memory deficits, and even neurodegeneration of cholinergic neurons.
The concept that endocytic alterations can promote AD is strengthened by this work, and by genetics studies implicating the endocytic system in AD. Genes regulating the endocytic pathway, such as rab5 and rab7, are upregulated in neurons from MCI and AD patients and abnormal endosomal enlargement is observed in early AD. Previous studies of sporadic AD brains showed that the volume of neuronal early endosomes was threefold larger than normal (Cataldo et al., 2000).
The endocytic pathway is responsible for the internalization and processing of cell-surface APP. Early endosomes are the first major sorting station on the endocytic pathway and the site of internalization and initial processing of proteins such as APP and BACE1. Inhibition of BACE1 is a therapeutic target to treat AD, however BACE1 inhibitors failed in clinical trials. BACE1 is involved in the proteolytic processing of other proteins with an important immunological function and it is essential to consider other strategies to selectively inhibit BACE1 cleavage of APP and not other substrates. BACE1 also cleaves the non-amyloid substrates NRG1 and L1 which are endocytosis independent.
Previously we proposed cell-surface APP as a target for immunotherapy of AD via antibodies directed to the β-secretase cleavage site of APP (BBS1). These antibodies didn’t recognize Aβ, reduced phosphorylated tau and tangles, and reduced levels of β-CTF (Arbel et al., 2005). The approach has an advantage over immunization strategies directed squarely at β-amyloid removal: Blocking Aβ production may allow the more gradual process of Aβ clearance to be better tolerated by the cerebral vasculature.
APP and BACE1 are internalized from the plasma membrane probably through separate routes, and merge at the early endosomal compartments. BACE1 can then be sorted to lysosomes for degradation or recycling to the cell surface. This approach demonstrates that inhibiting Aβ production via the endocytic pathway without affecting BACE1 processing of other substrates is possible and remains an obvious choice of clinical value for the specific treatment of AD. A similar approach was reported by the Rajendran group, showing that an endosomally targeted BACE1 inhibitor may spare NRG1 and L1 but inhibit APP processing (Ben Halima et al., 2016)
In landmark papers years ago, Nixon and colleagues described early, pre-plaque enlargement of Rab5 endosomes related to AD pathogenesis. Increased endocytosis will lead to more surface APP being internalized and trapped in the endocytic pathways, which in turn would result in enhancement of APP cleavage by BACE.
References:
Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000 Jul;157(1):277-86. PubMed.
Arbel M, Yacoby I, Solomon B. Inhibition of amyloid precursor protein processing by beta-secretase through site-directed antibodies. Proc Natl Acad Sci U S A. 2005 May 24;102(21):7718-23. PubMed.
Ben Halima S, Mishra S, Raja KM, Willem M, Baici A, Simons K, Brüstle O, Koch P, Haass C, Caflisch A, Rajendran L. Specific Inhibition of β-Secretase Processing of the Alzheimer Disease Amyloid Precursor Protein. Cell Rep. 2016 Mar 8;14(9):2127-41. Epub 2016 Feb 25 PubMed.
National Institutes of Health
This work points to the central role of early endocytosis in neurodegeneration. Studies of convergence of both GWAS hits and functional studies on early endocytosis, including this one, make it clear that disrupted early endocytosis is a key feature of neurodegeneration. Early endocytic defects have functional consequences beyond just their impact on APP or APP cleavage products.
This study reminds me of work reporting that APOE4 homozygous iPSC-derived neurons also have enlarged early endosomes (Lin et al., 2018), like those observed upon increasing Rab5 expression. Perhaps APOE4 here is having a similar phenotypic effect as increased Rab5 expression.
Interestingly, Lin et al. and our work (Narayan et al., 2020) has shown that, in the context of astrocytes, APOE4 results in a lower number of Rab5+ early endosomes. Overexpression of early endocytic factors like PICALM increases the efficiency of early endocytosis but, surprisingly, leads to compromised early endocytosis in a healthy APOE3 homozygous background. These and other studies make me very curious (1) whether different cell types respond the same way to increased Rab5 expression, and (2) whether increased expression of other early endocytic factors (in otherwise healthy contexts) may lead to similar detrimental consequences?
It is very possible that increasing Rab5 expression, as the authors did in this study, phenocopies the action of other risk factors aside from APOE4. This could suggest a further convergence of risk phenotypes for AD. There is clearly lots to look at in the future!
References:
Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, Rueda R, Kritskiy O, Abdurrob F, Peng Z, Milo B, Yu CJ, Elmsaouri S, Dey D, Ko T, Yankner BA, Tsai LH. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer's Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron. 2018 Jun 27;98(6):1141-1154.e7. Epub 2018 May 31 PubMed. Correction.
Narayan P, Sienski G, Bonner JM, Lin YT, Seo J, Baru V, Haque A, Milo B, Akay LA, Graziosi A, Freyzon Y, Landgraf D, Hesse WR, Valastyan J, Barrasa MI, Tsai LH, Lindquist S. PICALM Rescues Endocytic Defects Caused by the Alzheimer's Disease Risk Factor APOE4. Cell Rep. 2020 Oct 6;33(1):108224. PubMed.
View all comments by Priyanka NarayanEIP Pharma, Inc.
The Pensalfini article, taken together with a number of other recent articles, as well as long-standing scientific literature, helps pull together a number of emerging concepts toward two high-level, interrelated potential conclusions regarding the neurodegenerative process, particularly with respect to dementia:
(1) It argues for a disease pathogenesis model for dementia (AD and DLB) in which the core physiological defect that underlies the dementia (i.e., causes the cognitive deficits) is endolysosomal dysfunction;
(2) Endolysosomal dysfunction appears to be the mechanism by which tau pathology develops.
With regard to (1), the following points should be considered:
With regard to (2), we should consider:
All this speaks mostly to general dementia and neurodegeneration, and not to specific diseases, such as AD. Indeed, the nice aspect of the endolysosomal dysfunction model is that it can encompass dementia with Lewy bodies and other neurodegenerative diseases that have also been linked to Rab5 (see our review article). More specifically to AD, the results argue that a major component of disease pathogenesis that is a result of the FAD mutations in APP and PSEN1 appears to be driven by β-CTF-driven activation of Rab5. At the same time, the results do not preclude an effect of Aβ.
There are two components of AD that are not seen in the Rab5-overexpressing mouse: amyloid plaques and neurodegeneration in the hippocampus. The absence of amyloid plaques are easily explained—endogenous mouse Aβ does not form plaques due to differences in the physical chemistry between the mouse and human peptide. Also, while perhaps with aging there will be neurodegeneration in the hippocampus in these mice, it could also be that Aβ (or other components of APP metabolism) is involved in neurodegeneration in the hippocampus.
Finally, Pensalfini does not definitely exclude a role of Rab5 in increasing Aβ production, particularly in the context of other mutations that impact APP metabolism. Nor does it exclude the possibility that Aβ activates Rab5. In the end, the only way to know the relative contribution of the various potential effector pathways is to evaluate the clinical effects with treatments that target specific components of disease pathogenesis.
References:
Nixon RA. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer's disease: inseparable partners in a multifactorial disease. FASEB J. 2017 Jul;31(7):2729-2743. PubMed.
Xu W, Weissmiller AM, White JA 2nd, Fang F, Wang X, Wu Y, Pearn ML, Zhao X, Sawa M, Chen S, Gunawardena S, Ding J, Mobley WC, Wu C. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J Clin Invest. 2016 May 2;126(5):1815-33. Epub 2016 Apr 11 PubMed.
Germann UA, Alam JJ. P38α MAPK Signaling-A Robust Therapeutic Target for Rab5-Mediated Neurodegenerative Disease. Int J Mol Sci. 2020 Jul 31;21(15) PubMed.
Nuriel T, Peng KY, Ashok A, Dillman AA, Figueroa HY, Apuzzo J, Ambat J, Levy E, Cookson MR, Mathews PM, Duff KE. The Endosomal-Lysosomal Pathway Is Dysregulated by APOE4 Expression in Vivo. Front Neurosci. 2017;11:702. Epub 2017 Dec 12 PubMed.
Make a Comment
To make a comment you must login or register.