Earliest ALS Defects Said to Start in Disparate Places
Quick Links
Researchers have identified some of the earliest known defects in mouse models of amyotrophic lateral sclerosis, according to two papers in the January 14 Journal of Neuroscience. Signs of something amiss emerge within just a few weeks of birth in an aggressive model of ALS, report researchers from the University of Queensland in St. Lucia, Australia, who observed loss of dendritic spines in upper motor neurons. In a more slowly progressing model, the first signs of pathology occurred at the other end of the motor neuron network. Scientists from the University of Montréal in Canada found that in 4-month-old mice, the perisynaptic Schwann cells that surround neuromuscular junctions had trouble supporting synapse repair. Scientists hope that by understanding these precocious pathologies, they can come up with ways to slow down ALS from the get-go. That could help people who have a genetic predisposition for the disease.
Upper Motor Neuron Dendrites Wither Early
ALS, by definition, encompasses defects in both upper and lower motor neurons, though researchers have focused mostly on the latter. Lower motor neurons are hyperexcitable (Van Zundert et al., 2008), and Matthew Fogarty in Queensland, who works with senior authors Mark Bellingham and Peter Noakes, wanted to know if upper motor neurons were as well. He examined neural signaling in mice overexpressing human SOD1 with a glycine-93-alanine mutation. Disease moves fast in this model; spinal cord motor neurons start to degenerate at 1-2 months of age, and the mice develop noticeable symptoms, such as hind leg tremor, around 90 days after birth.
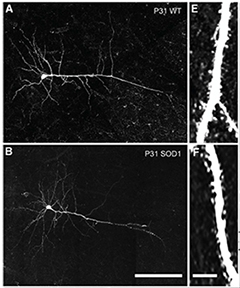
Disappearing Dendrites.
By just 1 month of age, the upper motor neuron dendrites of SOD1-G93A mice (bottom) have begun to shrink, compared with wild-type brains (top). [Image courtesy of Fogarty et al., The Journal of Neuroscience, 2015]
Fogarty observed more than twice as many excitatory neural signals coming into layer V cortical upper motor neurons in brain slices from 3-week-old SOD mice than in slices from wild-type animals. The dendritic spines had already begun to shrink at that age, and in slices from 4-week-old mice, the dendrites themselves had started to shorten (see image at right). “The cells are not dying yet, but they are definitely perturbed,” Fogarty said. These dendritic abnormalities mirror some of the earliest signs of decay in lower motor neurons, commented Andrew Eisen of the University of British Columbia in Vancouver, Canada, who was not involved in the study.
Synaptic Glia Ignore Synapses in Young SOD Mice
In Montréal, first author Danielle Arbour and colleagues examined the axons of lower motor neurons, where they innervate the neuromuscular junction. This synapse relies on support from local glia called perisynaptic Schwann cells, or PSCs. These have two modes, explained senior author Richard Robitaille. Normally, the PSCs are in maintenance mode. They sense the acetylcholine released by the presynaptic terminal and release factors that make the synapse efficient and stable. In cases of mild injury, partial denervation can occur, and the PSCs shift into repair mode. They sense and react to the reduction of acetylcholine, for example by removing debris so the synapse can re-form.
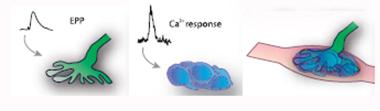
Junctions Gone Wrong.
In neuromuscular junctions, presynaptic signaling (left) activates PSCs (middle), which help maintain junction structure (right). This collaboration falters in ALS. [Image courtesy of Arbour et al., The Journal of Neuroscience, 2015.]
Several studies have suggested that astrocytes and microglia contribute to, and perhaps even instigate, neural death in ALS (see Apr 2007 news; Boillée et al., 2006). Arbour, Robitaille, and colleagues wondered if PSCs, too, might have a role. To catch early defects, they isolated nerve-muscle connections from a slow model of ALS, i.e., mice expressing SOD1 with the glycine-37-arginine mutation. In these animals, symptoms become noticeable at about 14 months of age, middle age for a mouse. However, at 4 months, Arbour saw presynaptic terminals firing more often and more strongly. At the same time, the PSCs became highly sensitive to acetylcholine. “To our knowledge, this is the earliest persistent change reported in this mouse model,” the authors wrote. In addition, by 13-months the mice’s junctions were disorganized, an indication that denervation had begun.
Robitaille hypothesized that the PSCs in the ALS mice are so hyper-responsive to acetylcholine that they do not notice when its concentration drops. Then, when ALS attacks the junctions, the PSCs fail to go into repair mode and the synapse deteriorates. “The perisynaptic Schwann cells are happy campers, blind to the problems [in ALS],” Robitaille said. The researchers next want to test their hypothesis in biopsy tissue from people with the disease.
“This is the first study I know to examine the involvement of perisynaptic Schwann cells in ALS,” commented Douglas Fields of the National Institute of Child Health and Human Development in Bethesda, Maryland (see full comment below). “It makes good sense that these cells would be involved in ALS and other neuromuscular disorders … these findings open up a new avenue of research into treatments that target these unusual and important glial cells.”
Early Onset
The evidence for problems in both kinds of motor neurons mimics human disease, which in Fogarty’s view highlights the importance of the mSOD1 mouse. “If we are going to treat ALS, we need to treat both lower motor neurons and upper motor neurons,” he said. Fogarty is now testing whether upper motor neurons respond to the same treatments as lower motor neurons do.
Many neurodegenerative diseases, including Alzheimer’s and Parkinson’s, get their start years or decades before people notice any symptoms (see Jan 2015 news). “In fact, they may begin at birth,” Eisen said. “There may be many years of metabolic abnormalities before any structural change” (see Eisen et al., 2014).
But where does ALS truly start? Many scientists subscribe to the “dying back” hypothesis, whereby degeneration begins at the neuromuscular junction when motor neurons retreat from the synapse (Fischer et al., 2004). A few others, such as Eisen, prefer the “dying forward” or upper motor neuron hypothesis. “We believe ALS begins in the brain,” before spreading to lower motor neurons, he explained. Hande Ozdinler of Northwestern University Feinberg School of Medicine in Chicago agreed (see full comment below). “Many new findings show that the cortex may actually be the starting point for defects,” wrote Ozdinler, who was not involved in the study. “We should begin to appreciate the importance of cortical components of motor neuron circuitry.”
The answer is important, Eisen points out, so clinicians know where to look for those first defects. However, there is no consensus on where disease starts. Carol Milligan of Wake Forest School of Medicine in Winston-Salem, North Carolina, was not so sure that there would turn out to be one point of origin. She studies lower motor neurons of week-old SOD1-G93A mice, and told Alzforum, “We could not distinguish something happening only at one site. It is probably a continuum.”—Amber Dance
References
News Citations
- Glia—Absolving Neurons of Motor Neuron Disease
- API Biomarker Data Mirror DIAN’s, Support Progression Models
Paper Citations
- van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH, Constantine-Paton M, Bellingham MC. Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci. 2008 Oct 22;28(43):10864-74. PubMed.
- Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006 Oct 5;52(1):39-59. PubMed.
- Eisen A, Kiernan M, Mitsumoto H, Swash M. Amyotrophic lateral sclerosis: a long preclinical period?. J Neurol Neurosurg Psychiatry. 2014 Mar 19; PubMed.
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004 Feb;185(2):232-40. PubMed.
Further Reading
Papers
- Thomsen GM, Gowing G, Latter J, Chen M, Vit JP, Staggenborg K, Avalos P, Alkaslasi M, Ferraiuolo L, Likhite S, Kaspar BK, Svendsen CN. Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex. J Neurosci. 2014 Nov 19;34(47):15587-600. PubMed.
- Jara JH, Villa SR, Khan NA, Bohn MC, Ozdinler PH. AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol Dis. 2012 Aug;47(2):174-83. PubMed.
- Pieri M, Carunchio I, Curcio L, Mercuri NB, Zona C. Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp Neurol. 2009 Feb;215(2):368-79. PubMed.
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000 Apr 1;20(7):2534-42. PubMed.
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006 Mar;9(3):408-19. PubMed.
- Dupuis L, Gonzalez de Aguilar JL, Echaniz-Laguna A, Eschbach J, Rene F, Oudart H, Halter B, Huze C, Schaeffer L, Bouillaud F, Loeffler JP. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS One. 2009;4(4):e5390. PubMed.
Primary Papers
- Arbour D, Tremblay E, Martineau É, Julien JP, Robitaille R. Early and persistent abnormal decoding by glial cells at the neuromuscular junction in an ALS model. J Neurosci. 2015 Jan 14;35(2):688-706. PubMed.
- Fogarty MJ, Noakes PG, Bellingham MC. Motor Cortex Layer V Pyramidal Neurons Exhibit Dendritic Regression, Spine Loss, and Increased Synaptic Excitation in the Presymptomatic hSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. J Neurosci. 2015 Jan 14;35(2):643-7. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Northwestern University, Feinberg School of Medicine
The paper is succinct and reveals an important message. I want to applaud the authors for revealing very early spine defects in the apical dendrites of corticospinal motor neurons.
There is growing evidence revealing the importance of corticospinal motor neurons (CSMN) to disease pathology in ALS, and this paper shows that spine density and morphology changes occur as early as 21 days old, and that there potentially could be cortical connectivity defects, as well. This is of great interest because we were told for many years that the upper motor neurons were not critical for ALS pathology, or that their degeneration was secondary to spinal motor neuron loss. However, many new findings show that the cortex may actually be the starting point for defects in the motor neuron circuitry.
This paper builds upon previous findings emerging from our lab and from others suggesting that upper motor neuron degeneration is an early event in ALS. It is very important that they show the cellular defects occur at a very early age, even before the spinal motor neuron loss occurs. While this brief communication may not be strong enough to change the field, it is strong enough to add one more twist—it is now even more interesting to find the intrinsic and extrinsic factors that contribute to CSMN vulnerability, and how they relate to disease pathology.
That motor neuron death is early and not secondary to spinal motor neuron loss is a very important concept. We should begin to appreciate the importance of the cortex and the cortical component of motor neuron circuitry. The tools, reporter lines, and animal models to investigate this neuron population in detail are now available. It will be interesting to see if ALS could initiate from the cortex after all.
View all comments by P. Hande OzdinlerNIH/NICHD
This is a very interesting paper from the laboratory of one of the foremost experts on interactions between perisynaptic Schwann cells and neurotransmission at the neuromuscular junction. This is the latest in a series of important studies showing the involvement of glial cells in ALS. ALS, like most neurodegenerative diseases, was once believed to be a strictly neuronal disorder, but today we know that all types of glial cells, astrocytes, oligodendrocytes, Schwann cells, microglia, and now perisynaptic (terminal) Schwann cells are involved.
This is the first study I know to examine the involvement of perisynaptic Schwann cells in ALS. These cells surround the neuromuscular junction much like astrocytes surround synapses in the brain and spinal cord, and they seem to have many of the same functions. Perisynaptic Schwann cells modulate the strength of neurotransmission with muscle fibers, and they are especially important in remodeling the neuromuscular junction and in repairing it after injury. It makes good sense that these cells would be involved in ALS and other neuromuscular disorders.
These findings open a new avenue of research for developing new treatments for ALS that target these unusual and important glial cells.
View all comments by R. Douglas FieldsUniversity of Sydney
Where does ALS begin? Despite Charcot's initial observation of concomitant upper (UMN) and lower motor neuron (LMN) pathological changes in ALS, the question of where ALS begins has not been definitively established. Resolution of this question would enhance understanding of the pathophysiology of ALS, with diagnostic and therapeutic importance.
The dying-forward hypothesis proposes that ALS is mainly a disorder of corticomotoneurons, which connect monosynaptically with anterior horn cells, mediating anterograde degeneration of anterior horn cells via glutamate excitotoxicity. Support for a dying-forward hypothesis includes results from transcranial magnetic stimulation studies documenting that cortical hyperexcitability is an early feature in patients with sporadic ALS and precedes the clinical onset of familial ALS.
Clinical support comes from observations that motor neurons without a monosynaptic connection with corticomotoneurons, such as the oculomotor, abducens, and Onuf's nuclei, are typically spared in ALS; and that pure LMN forms of ALS are rare, whereas subclinical UMN involvement is invariably detected.
Further support has now been provided by Bellingham and colleagues, who identify changes consistent with very early degeneration of the upper motor neuron in the SOD1 mouse model, with loss of dendritic spines at 3 weeks. These changes are associated with the development of hyperexcitability and the advent of neurodegeneration.
While these animal studies are now consistent with clinical findings and experimental observations, an ongoing concern relates to the suitability of the SOD1 model for the human disease of ALS. Until recently, this mouse model was considered by some as the benchmark for testing potential neuroprotectants in ALS. Unfortunately the positive results from trials undertaken in the SOD1 mouse model have not translated to therapy for patients diagnosed with ALS.
Why has this mismatch occurred? In part, given that SOD1 mutations account for about 2 percent of all ALS cases, it may be argued that this model might have less relevance to human sporadic disease. Furthermore, the SOD1 model undergoes a series of stereotypical changes that begin with hind limb weakness.
In terms of future approaches, strategies aimed at modulating gene expression are now emerging as potential novel therapeutic options, particularly in light of significant advances in the understanding of the genetic causes of ALS. One such approach involves the use of antisense oligonucleotides which, when delivered intrathecally, reduce mRNA and SOD1 protein concentrations in brain and spinal cord in the SOD1 mouse and prolongs survival.
Separately, the recent development of mouse models with mutations in the gene encoding TDP-43 is a further potential advance in therapeutic development for ALS, providing basic scientists with a new, perhaps more relevant, platform for studying novel therapies.
View all comments by Matthew KiernanUniversity of Southern California
This paper by Arbour et al. from the Robitaille lab reports for the first time that glial cells called perisynaptic Schwann cells (PSCs, aka terminal Schwann cells) at the neuromuscular junction (NMJ) show abnormal properties at the presymptomatic and pre-onset stages in a slowly progressive mouse model (SOD1G37R) of amyotrophic lateral sclerosis (ALS). It is thought that NMJ dysfunction contributes to the pathogenesis of ALS. Given that PSCs are essential to synaptic maintenance and repair at the NMJ, it is critical to examine whether PSC dysfunction may contribute to NMJ dysfunction in this disease. This carefully executed study is very important as it further demonstrates the key role of PSCs in synaptic modulation, maintenance, and repairs at the NMJ, not only in health but also in disease. This study also provides a novel concept of ALS disease mechanisms involving peripheral glial cells.
This paper also raises a number of interesting questions for future investigation. For example, what triggers the enhanced intracellular Ca2+ activation in PSCs and the increased transmitter release seen in these mutant mice? Whether and how does the Ca2+ enhancement play a direct role in NMJ dysfunction in ALS? What happens to PSCs and synaptic strength at later symptomatic stages? Do the same changes occur in other ALS mouse models, such as SOD1G93A, which has a relatively earlier disease onset and is thought to involve dying back of motor nerve terminal at the NMJ? Could activation of G protein pathways in mutant PSCs modulate transmitter release in ALS mice, in a way similar to their previous findings at the normal NMJ? And how can the inadequate activation of muscarinic acetylcholine receptors seen in mutant PSCs be corrected, and could such correction could be used for potential ALS therapy?
Further studies on these questions and the mechanisms of the alterations of the intrinsic PSC properties and synaptic strength would provide a better understanding of NMJ dysfunction in ALS and may lead to a potential therapy by targeting these Schwann cells.
View all comments by Chien-Ping KoNorthwestern University
Northwestern University
Nothwestern University Feinberg School of Medicine
Cortical abnormalities in hSOD1G93A mouse model confirm a key role of synaptic dysfunction in ALS
Amyotrophic lateral sclerosis (ALS) is characterized by the concomitant dysfunction of corticospinal motoneurons (CSMNs) in the primary motor cortex, and motoneurons of the spinal cord and brainstem that CSMNs activate. Postmortem examination of CSMNs in patients after Golgi staining reveals vast morphological abnormalities, including stunted basal dendritic arborization, gnarled apical dendrites, and spine loss (Hammer et al., 1979). Furthermore, CSMN hyperexcitability has been detected in sporadic ALS patients and in presymptomatic familial ALS patients (Eisen et al., 1993; Vucic et al., 2008). These and other studies suggest that loss of voluntary motor function in ALS patients is initiated by cortical dysfunction (hyperexcitability of CSMNs), and subsequently spreads to spinal motoneurons (Braak et al., 2013). Moreover, one theory on why some spinal motoneurons (SMNs) are selectively vulnerable in ALS while others are completely unaffected focuses on the CSMNs and their downstream synaptic targets. The subsets of motoneurons which are innervated by CSMNs in humans are generally most vulnerable to degeneration in ALS patients, while those with little CSMN input are spared (Eisen et al., 1992). Hence it has been proposed that synaptic connectivity from the CSMNs could be the key to selective vulnerability in ALS.
It is therefore puzzling that in the hSOD1G93A mouse model of ALS, dysfunction in SMNs has been more thoroughly characterized than in CSMNs. That is, until recently: Several exciting studies have now revealed insights into early pathological events in CSMNs. The first report, in 2002, showed a loss of corticospinal projections in the hSOD1G93A mouse before overt symptom onset (projections lost by postnatal day [P] 60, while tremor and weakness appear in hindlimbs around P90) (Zang and Cheema, 2002). Then, in 2009, electrophysiological abnormalities in cultured embryonic cortical neurons were described by the Zona laboratory at the University of Rome (Pieri et al., 2009). Coincidentally, electrical changes in cultured cortical neurons mirrored many of the changes observed in cultured embryonic SMNs, including increased firing frequencies of action potentials and a larger-amplitude persistent inward current (Pieri et al., 2003; Kuo et al., 2004; Kuo et al., 2005). In addition to those abnormalities, studies of hSOD1G93A brainstem and spinal motoneurons have shown altered dendritic arborization (reduced in embryonic spinal and postnatal brainstem motoneurons, and expanded postnatally in spinal neurons) (Martin et al., 2013; van Zundert et al., 2008; Amendola and Durand, 2008; Filipchuk and Durand, 2012), increased persistent inward currents throughout development (Kuo et al., 2005; van Zundert et al., 2008; Quinlan et al., 2011; Delestree et al., 2014), and eventual progression of motoneurons to hypoexcitability just preceding symptom onset (Delestree et al., 2014). Additional dysfunction has been documented presymptomatically in axon transport (Bilsland et al., 2010; De Vos et al., 2007; Williamson and Cleveland, 1999; Zhang et al., 1997; Kieran et al., 2005; Warita et al., 1999), mitochondrial function (Li et al., 2010; Jaiswal and Keller, 2009; Mattiazzi et al., 2002; Damiano et al., 2006; Nguyen et al., 2009; Bilsland et al., 2008), protein handling and ER stress (Saxena et al., 2009), all in brainstem and spinal motoneurons. Research on the pathophysiology of CSMNs is finally catching up. Recent studies now show that, presymptomatically, CSMNs are just as perturbed as spinal motoneurons, if not more so. Ozdinler and colleagues at Harvard and Northwestern demonstrate degeneration in the apical dendrite of CSMNs, decreased spine density, and decreased numbers of CSMNs from P60 (Jara et al., 2012; Ozdinler et al., 2011).
Now, further inroads are reported by Mark Bellingham's laboratory at the University of Queensland. Fogarty and colleagues demonstrate an even earlier onset of pathology in layer V pyramidal neurons, a group of cortical neurons that includes CSMNs. The authors show apical dendrite degeneration starting from P28, and even earlier spine loss along the apical dendrite, from P21. Anatomical changes in the spines are likely driven synaptically, and Fogarty et al. give electrophysiological evidence of this: Spine loss is accompanied by an increased frequency of excitatory postsynaptic currents (EPSCs) in layer V pyramidal neurons (Fogarty et al., 2015). These results constitute the first evidence of a disturbance in the cortical circuitry in ALS starting at P21, an age at which the mice are just weaned, long before symptom onset.
Another study out last month in Cerebral Cortex from the Zona laboratory (Saba et al., 2015) presented other clues to the overactive EPSCs. In addition to showing the same increased frequency of EPSCs in layer V pyramidal neurons in hSOD1G93A mice at P26-31, they showed EPSCs were mediated via non-NMDA type glutamate receptors, and even when action potential-mediated neurotransmission was blocked, an increased frequency in mini EPSCs remained. Through a combination of immunohistochemistry, western blotting, and quantitative PCR, they demonstrated enhanced vesicular glutamate transporter 2 (VGlut2) expression. Specifically, more glutamatergic contacts formed on layer V pyramidal neurons in layer II/III and V of primary motor cortex. Thalamocortical projections to layer II/III and V of the motor cortex are VGlut2+ and are a potential source of the EPSCs barraging layer V pyramidal neurons (Kuramoto et al., 2009).
The evidence suggests that instead of cortical neurons initiating symptoms of ALS, they could just be on the receiving end of faulty synaptic drive. Rather than vulnerable neuronal populations explaining ALS, perhaps the problem is vulnerable circuits. It would be interesting to see future studies probe deeper into possible dysfunction in the cortical circuitry in ALS. Meanwhile, advances in the pathophysiology of brainstem and spinal motoneurons in ALS also lead to questions of whether the circuitry is perturbed there as well (van Zundert et al., 2008; Saxena et al., 2013; Bories et al., 2007; Vinsant et al., 2013; Vinsant et al., 2013; Casas et al., 2013; Martin et al., 2007; Chang and Martin, 2009; Wootz et al., 2013).
Interestingly, even though the hSOD1G93A mouse faithfully recapitulates numerous features of ALS, including CSMN degeneration, muscle weakness, muscle atrophy, spinal motor neuron loss and progressive paralysis (Gurney, 1994), a valid criticism of this model is that rodents generally lack direct synaptic inputs from CSMNs to SMNs (Alstermark et al., 2004; Yang and Lemon, 2003). However, with concomitant degeneration now demonstrated in both CSMNs and SMNs in the mouse model, this may indicate that these specific synaptic connections are less important in conferring dysfunction than overall homeostasis of the motor control network. To support this, the sole FDA-approved drug treatment for ALS, riluzole, seems to exert its beneficial effects both by dampening cellular excitability and network excitability (Bellingham, 2011; Miller et al., 2012). Although the hSOD1G93A mouse is not a perfect model of ALS, it remains a useful tool, as these studies demonstrate. This model, along with more recently developed animal models of ALS using mutations in the UBQLN2 (Gorrie et al., 2014), TARDP (Wegorzewska et al., 2009; Wils et al., 2010; Xu et al., 2010) and FUS/TLS (Huang et al., 2011) genes, as well as induced pluripotent stem cells from ALS patients (Dimos et al., 2008; Devlin et al., 2015), provide us with the means to study the mechanisms of disease in ALS at all levels, from individual cells to neural circuits, in search of more accessible and effective targets for treatment.
References:
Hammer RP Jr, Tomiyasu U, Scheibel AB. Degeneration of the human Betz cell due to amyotrophic lateral sclerosis. Exp Neurol. 1979 Feb;63(2):336-46. PubMed.
Eisen A, Pant B, Stewart H. Cortical excitability in amyotrophic lateral sclerosis: a clue to pathogenesis. Can J Neurol Sci. 1993 Feb;20(1):11-6. PubMed.
Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain. 2008 Jun;131(Pt 6):1540-50. Epub 2008 May 9 PubMed.
Braak H, Brettschneider J, Ludolph AC, Lee VM, Trojanowski JQ, Del Tredici K. Amyotrophic lateral sclerosis--a model of corticofugal axonal spread. Nat Rev Neurol. 2013 Dec;9(12):708-14. Epub 2013 Nov 12 PubMed.
Eisen A, Kim S, Pant B. Amyotrophic lateral sclerosis (ALS): a phylogenetic disease of the corticomotoneuron?. Muscle Nerve. 1992 Feb;15(2):219-24. PubMed.
Zang DW, Cheema SS. Degeneration of corticospinal and bulbospinal systems in the superoxide dismutase 1(G93A G1H) transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett. 2002 Oct 31;332(2):99-102. PubMed.
Pieri M, Carunchio I, Curcio L, Mercuri NB, Zona C. Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp Neurol. 2009 Feb;215(2):368-79. PubMed.
Pieri M, Albo F, Gaetti C, Spalloni A, Bengtson CP, Longone P, Cavalcanti S, Zona C. Altered excitability of motor neurons in a transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett. 2003 Nov 20;351(3):153-6. PubMed.
Kuo JJ, Schonewille M, Siddique T, Schults AN, Fu R, Bär PR, Anelli R, Heckman CJ, Kroese AB. Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J Neurophysiol. 2004 Jan;91(1):571-5. PubMed.
Kuo JJ, Siddique T, Fu R, Heckman CJ. Increased persistent Na(+) current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol. 2005 Mar 15;563(Pt 3):843-54. PubMed.
Martin E, Cazenave W, Cattaert D, Branchereau P. Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2013 Jun;54:116-26. PubMed.
van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH, Constantine-Paton M, Bellingham MC. Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci. 2008 Oct 22;28(43):10864-74. PubMed.
Amendola J, Durand J. Morphological differences between wild-type and transgenic superoxide dismutase 1 lumbar motoneurons in postnatal mice. J Comp Neurol. 2008 Nov 20;511(3):329-41. PubMed.
Filipchuk AA, Durand J. Postnatal dendritic development in lumbar motoneurons in mutant superoxide dismutase 1 mouse model of amyotrophic lateral sclerosis. Neuroscience. 2012 May 3;209:144-54. Epub 2012 Feb 11 PubMed.
Quinlan KA, Schuster JE, Fu R, Siddique T, Heckman CJ. Altered postnatal maturation of electrical properties in spinal motoneurons in a mouse model of amyotrophic lateral sclerosis. J Physiol. 2011 May 1;589(Pt 9):2245-60. Epub 2011 Feb 28 PubMed.
Delestrée N, Manuel M, Iglesias C, Elbasiouny SM, Heckman CJ, Zytnicki D. Adult spinal motoneurones are not hyperexcitable in a mouse model of inherited amyotrophic lateral sclerosis. J Physiol. 2014 Apr 1;592(Pt 7):1687-703. Epub 2014 Jan 20 PubMed.
Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G. Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci U S A. 2010 Nov 23;107(47):20523-8. Epub 2010 Nov 8 PubMed.
De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CC, Grierson AJ. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007 Nov 15;16(22):2720-8. Epub 2007 Aug 28 PubMed.
Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999 Jan;2(1):50-6. PubMed.
Zhang B, Tu P, Abtahian F, Trojanowski JQ, Lee VM. Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J Cell Biol. 1997 Dec 1;139(5):1307-15. PubMed.
Kieran D, Hafezparast M, Bohnert S, Dick JR, Martin J, Schiavo G, Fisher EM, Greensmith L. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J Cell Biol. 2005 May 23;169(4):561-7. PubMed.
Warita H, Itoyama Y, Abe K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1999 Feb 20;819(1-2):120-31. PubMed.
Li Q, Vande Velde C, Israelson A, Xie J, Bailey AO, Dong MQ, Chun SJ, Roy T, Winer L, Yates JR, Capaldi RA, Cleveland DW, Miller TM. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc Natl Acad Sci U S A. 2010 Dec 7;107(49):21146-51. PubMed.
Jaiswal MK, Keller BU. Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol Pharmacol. 2009 Mar;75(3):478-89. Epub 2008 Dec 5 PubMed.
Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002 Aug 16;277(33):29626-33. PubMed.
Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, Flint Beal M, Manfredi G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem. 2006 Mar;96(5):1349-61. PubMed.
Nguyen KT, García-Chacón LE, Barrett JN, Barrett EF, David G. The Psi(m) depolarization that accompanies mitochondrial Ca2+ uptake is greater in mutant SOD1 than in wild-type mouse motor terminals. Proc Natl Acad Sci U S A. 2009 Feb 10;106(6):2007-11. PubMed.
Bilsland LG, Nirmalananthan N, Yip J, Greensmith L, Duchen MR. Expression of mutant SOD1 in astrocytes induces functional deficits in motoneuron mitochondria. J Neurochem. 2008 Dec;107(5):1271-83. Epub 2008 Oct 25 PubMed.
Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009 May;12(5):627-36. PubMed.
Jara JH, Villa SR, Khan NA, Bohn MC, Ozdinler PH. AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol Dis. 2012 Aug;47(2):174-83. PubMed.
Ozdinler PH, Benn S, Yamamoto TH, Güzel M, Brown RH, Macklis JD. Corticospinal motor neurons and related subcerebral projection neurons undergo early and specific neurodegeneration in hSOD1G⁹³A transgenic ALS mice. J Neurosci. 2011 Mar 16;31(11):4166-77. PubMed.
Fogarty MJ, Noakes PG, Bellingham MC. Motor Cortex Layer V Pyramidal Neurons Exhibit Dendritic Regression, Spine Loss, and Increased Synaptic Excitation in the Presymptomatic hSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. J Neurosci. 2015 Jan 14;35(2):643-7. PubMed.
Saba L, Viscomi MT, Caioli S, Pignataro A, Bisicchia E, Pieri M, Molinari M, Ammassari-Teule M, Zona C. Altered Functionality, Morphology, and Vesicular Glutamate Transporter Expression of Cortical Motor Neurons from a Presymptomatic Mouse Model of Amyotrophic Lateral Sclerosis. Cereb Cortex. 2015 Jan 16; PubMed.
Kuramoto E, Furuta T, Nakamura KC, Unzai T, Hioki H, Kaneko T. Two types of thalamocortical projections from the motor thalamic nuclei of the rat: a single neuron-tracing study using viral vectors. Cereb Cortex. 2009 Sep;19(9):2065-77. Epub 2009 Jan 27 PubMed.
Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F, Caroni P. Neuroprotection through Excitability and mTOR Required in ALS Motoneurons to Delay Disease and Extend Survival. Neuron. 2013 Oct 2;80(1):80-96. PubMed.
Bories C, Amendola J, Lamotte d'Incamps B, Durand J. Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci. 2007 Jan;25(2):451-9. PubMed.
Vinsant S, Mansfield C, Jimenez-Moreno R, Del Gaizo Moore V, Yoshikawa M, Hampton TG, Prevette D, Caress J, Oppenheim RW, Milligan C. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part I, background and methods. Brain Behav. 2013 Jul;3(4):335-50. Epub 2013 Jun 11 PubMed.
Casas C, Herrando-Grabulosa M, Manzano R, Mancuso R, Osta R, Navarro X. Early presymptomatic cholinergic dysfunction in a murine model of amyotrophic lateral sclerosis. Brain Behav. 2013 Mar;3(2):145-58. Epub 2013 Feb 17 PubMed.
Martin LJ, Liu Z, Chen K, Price AC, Pan Y, Swaby JA, Golden WC. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J Comp Neurol. 2007 Jan 1;500(1):20-46. PubMed.
Chang Q, Martin LJ. Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice: a quantitative confocal analysis. Am J Pathol. 2009 Feb;174(2):574-85. Epub 2008 Dec 30 PubMed.
Wootz H, Fitzsimons-Kantamneni E, Larhammar M, Rotterman TM, Enjin A, Patra K, André E, Van Zundert B, Kullander K, Alvarez FJ. Alterations in the motor neuron-renshaw cell circuit in the Sod1(G93A) mouse model. J Comp Neurol. 2013 May 1;521(7):1449-69. PubMed.
Gurney ME. Transgenic-mouse model of amyotrophic lateral sclerosis. N Engl J Med. 1994 Dec 22;331(25):1721-2. PubMed.
Alstermark B, Ogawa J, Isa T. Lack of monosynaptic corticomotoneuronal EPSPs in rats: disynaptic EPSPs mediated via reticulospinal neurons and polysynaptic EPSPs via segmental interneurons. J Neurophysiol. 2004 Apr;91(4):1832-9. Epub 2003 Nov 5 PubMed.
Yang HW, Lemon RN. An electron microscopic examination of the corticospinal projection to the cervical spinal cord in the rat: lack of evidence for cortico-motoneuronal synapses. Exp Brain Res. 2003 Apr;149(4):458-69. Epub 2003 Feb 21 PubMed.
Bellingham MC. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade?. CNS Neurosci Ther. 2011 Feb;17(1):4-31. PubMed.
Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012 Mar 14;3:CD001447. PubMed.
Gorrie GH, Fecto F, Radzicki D, Weiss C, Shi Y, Dong H, Zhai H, Fu R, Liu E, Li S, Arrat H, Bigio EH, Disterhoft JF, Martina M, Mugnaini E, Siddique T, Deng HX. Dendritic spinopathy in transgenic mice expressing ALS/dementia-linked mutant UBQLN2. Proc Natl Acad Sci U S A. 2014 Oct 7;111(40):14524-9. Epub 2014 Sep 22 PubMed.
Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009 Nov 3;106(44):18809-14. Epub 2009 Oct 15 PubMed.
Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2010 Feb 23;107(8):3858-63. Epub 2010 Feb 3 PubMed.
Xu YF, Gendron TF, Zhang YJ, Lin WL, D'Alton S, Sheng H, Casey MC, Tong J, Knight J, Yu X, Rademakers R, Boylan K, Hutton M, McGowan E, Dickson DW, Lewis J, Petrucelli L. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010 Aug 11;30(32):10851-9. PubMed.
Huang C, Zhou H, Tong J, Chen H, Liu YJ, Wang D, Wei X, Xia XG. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011 Mar;7(3):e1002011. PubMed.
Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008 Aug 29;321(5893):1218-21. PubMed.
Devlin AC, Burr K, Borooah S, Foster JD, Cleary EM, Geti I, Vallier L, Shaw CE, Chandran S, Miles GB. Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat Commun. 2015 Jan 12;6:5999. PubMed.
View all comments by Katharina QuinlanMake a Comment
To make a comment you must login or register.