Endoplasmic Reticulum Protein Protects Motor Neurons from ALS
Quick Links
When amyotrophic lateral sclerosis attacks motor neurons, certain types resist while others surrender. If scientists can figure out why some neurons hang tough, they may be able to offer that same protection to those that succumb. Smita Saxena and colleagues at the University of Bern in Switzerland believe the endoplasmic reticulum helps explain the selective vulnerability. In the January 5 Nature Neuroscience online, they report that the ALS-resistant motor neurons draw defense from a protein called Sil1, which is in short supply in the sensitive neurons. “At the level of the ER, our recent data suggests that Sil1 is one of the main players,” Saxena said. Homozygous mutations in the SIL1 gene cause muscle weakness in children, while studies hint that heterozygous Sil1 mutations might contribute to ALS risk. Saxena does not know how Sil1 shores up the ER of the resistant neurons, or how it protects them.
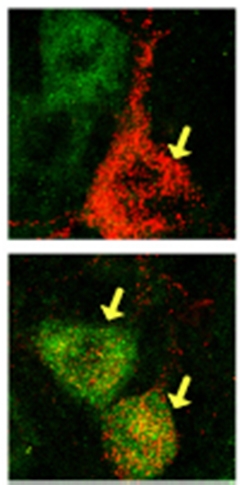
Sil1 Silos.
Motor neurons susceptible to ALS (top, red) contain little Sil1 (green) compared to resistant ones (bottom). [Image courtesy of Filézac de L’Etang et al., Nature Neuroscience, University of Bern.]
In ALS model mice, the first to succumb are the fast-fatigable motor neurons needed for rapid motions such as jumping. In contrast, slow motor neurons, responsible for long-term activities like standing, stay connected to muscle and survive until the animal’s death. In a previous study, Saxena discovered that the vulnerable motor neurons upregulate markers for ER stress, turning on a pathway the cell uses to deal with misfolded proteins (see Mar 2009 news). While a short-term version of this unfolded protein response protects cells from malformed peptides, Saxena thinks chronic activation makes the neurons sicker.
To investigate what keeps the ER of slow motor neurons from stressing out, first author Audrey Filézac de L’Etang and colleagues isolated fast-fatigable and slow motor neurons that innervate leg muscles of wild-type mice. With quantitative polymerase chain reaction, they measured the levels of various mRNAs that encode ER-based proteins. Most were similarly abundant in each neuron type. Sil1 stood out because the slow motor neurons contained more than six times as much of it as the fast-fatigable ones.
Sil1 acts as a co-chaperone that helps another ER resident, binding immunoglobulin protein (BiP), in its job as chaperone and sensor for ER stress. Homozygous loss-of-function mutations in Sil1 cause Marinesco-Sjögren syndrome. This pediatric condition causes symptoms such as muscle weakness and uncoordinated movement, as well as vision problems and intellectual disability. ALS-like motor neuron disease has not typically been reported in people with Marinesco-Sjögren syndrome, however, Saxena noted that physicians tend to examine patients most thoroughly when they are younger, perhaps before motor neuron problems might develop.
Genetic data back up the link between Sil1 and ALS. Genome-wide association studies have identified a dozen nucleotide swaps in Sil1 in people with ALS (see ALSGene). Saxena speculated that while full loss of function mutations in Sil1 cause Marinesco-Sjögren syndrome, milder sequence changes might be a risk factor for ALS. Also supporting a role for Sil1 in motor neurons, Filézac de L’Etang and co-authors found that the neuromuscular junctions of so-called woozy mice, which lack Sil1, exhibited several defects. Axons filled up with autophagic vacuoles and separated from the muscles, causing muscle atrophy.
To determine how Sil1 influences ALS progression, Filézac de L’Etang crossed the Sil1 mutants with mice carrying human mutant SOD1 to make ALS mice with only one copy of Sil1. ALS model mice vary in SOD1 copy number; in these experiments, the authors used a low-copy-number version that survived an average of 284 days. Those with only one copy of Sil1 lasted for just 248 days. The double mutants struggled to balance on a rotating rod at a younger age than mSOD1 controls.
If less Sil1 makes the mice ill, could extra Sil1 help them out? When the researchers delivered the Sil1 gene by way of adenovirus to the motor neurons of mSOD1 mice, it delayed both unfolded protein response and axon degeneration in fast-fatigable motor neurons. The mice were stronger and more coordinated than untreated mSOD1 mice on the rotarod test, and lived longer. ALS mice with high levels of the transgene gained a month; those with moderate mSOD1 expression gained nearly three.
The mechanism by which Sil1 protects motor neurons remains fuzzy. It might carry multiple, parallel protective actions, Saxena speculated. In N2a neuroblastoma cultures, Sil1 reduced the accumulation of misfolded SOD1. BiP must be involved in this process, because when Filezac de L’Etang mutated Sil1’s BiP binding site, SOD1 built up again. Sil1 overexpression also upregulated ER chaperones that are normally downregulated in mSOD1 neurons, the authors found. “This whole process somehow changes the milieu of the ER,” Saxena said, explaining that the change makes the organelle “fitter” and able to keep operating without turning on the unfolded protein response. That would explain why slow motor neurons, with high Sil1 levels, tend to withstand the onslaught of ALS, while fast-fatigable motor neurons quickly turn to the unfolded protein response, which they cannot maintain long enough to survive.
Not all scientists believe that ER stress contributes significantly to ALS. Serge Przedborski of Columbia University in New York, who was not involved in the work, pointed out that because ALS mice express extra copies of the SOD1 transgene, they pump out much more toxic protein than the neurons of a person with the disease. Even the low-copy-number SOD1 mouse, which Saxena and colleagues used to address this concern, has up to 10 SOD1 genes. The ER stress response would be a natural reaction to such a high concentration of any faulty protein, Przedborski suggested, but that might never occur in people.
Arthur Horwich of the Yale University School of Medicine in New Haven, Connecticut, who did not participate in the study, also questioned the importance of the ER in ALS in an email to Alzforum (see full comment below). His group has not observed ER stress in mice toting a SOD1-glycine-85-arginine mutation, different from the glycine-93-alanine model used by the Saxena lab. “I worry that ER stress is not a primary and consistent ALS-related pathology,” Horwich wrote. Saxena noted that in her 2009 study, she saw markers for ER stress in the transcriptome of fast-fatigable motor neurons from SOD1-G85R animals. In the current work, the authors observed mislocalized and aggregated Sil1 in mice overexpressing another ALS gene, TDP-43, and in spinal cord samples from people who died of sporadic ALS. These data were correlative and do not prove a role for Sil1 in those cases, Przedborski said.—Amber Dance
References
News Citations
External Citations
Further Reading
Papers
- Prell T, Lautenschläger J, Witte OW, Carri MT, Grosskreutz J. The unfolded protein response in models of human mutant G93A amyotrophic lateral sclerosis. Eur J Neurosci. 2012 Mar;35(5):652-60. PubMed.
- Walker AK, Atkin JD. Stress signaling from the endoplasmic reticulum: A central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB Life. 2011 Aug 10; PubMed.
- Sasaki S. Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2010 Apr;69(4):346-55. PubMed.
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009 May;12(5):627-36. PubMed.
- Kanekura K, Suzuki H, Aiso S, Matsuoka M. ER stress and unfolded protein response in amyotrophic lateral sclerosis. Mol Neurobiol. 2009 Apr;39(2):81-9. PubMed.
- Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008 Jun;30(3):400-7. PubMed.
News
- Surprise Save: Excitability Protects Neurons from Lou Gehrig’s
- No MAM: ALS Protein Breaks Mitochondria-Endoplasmic Reticulum Bond
- As Calcium Control Fades, So Do Motor Neurons in ALS Model
- New ALS Genes Implicate Protein Degradation, Endoplasmic Reticulum
- Honolulu: Potential New Gene on the ALS List
Primary Papers
- Filézac de L'Etang A, Maharjan N, Cordeiro Braña M, Ruegsegger C, Rehmann R, Goswami A, Roos A, Troost D, Schneider BL, Weis J, Saxena S. Marinesco-Sjögren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat Neurosci. 2015 Feb;18(2):227-38. Epub 2015 Jan 5 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
This is a provocative paper that follows on from the observation that the unfolded protein response of the endoplasmic reticulum (ER) becomes activated in a fraction of motor neurons at a point in the progression of some forms of ALS. At the circuit level, the idea of selective expression of Sil1 in specific motor neuron firing types (slow-firing), and also of other components (MMP9 was reported in fast-firing MNs), emphasizes our need to understand what it is that makes FF motor neurons selectively vulnerable. It may be a multiplicity of factors. Whatever the reason, it seems like overexpression of Sil1 increases longevity of SOD G93A animals.
While we would agree that fast-firing motor neurons are the vulnerable ones (see Hadzipasic et al., 2014), we see no ER stress in the G85R SOD1YFP model in motor neurons (Bandyopadhyay et al, 2013), but investigators studying G93A always seem to see evidence of ER stress. I wonder if it might be secondary to having free metals fall off of G93A, since the protein is constantly folding and unfolding. Under the electron microscope, membranes in G93A seem damaged, perhaps due to free radicals, whereas in G85R (which never folds and does not appear to take on metals) the organellar and other membranes look intact. I worry that ER stress is not a primary and consistent ALS-related pathology, or at least the G85R SOD1YFP fusion animals do not require it in order to go on to paralysis. Nonetheless, this study seems to have a fair-sized effect on survival of G93A by altering Sil1.
Mechanistically, one could understand that overexpression of this nucleotide exchanger of BiP at the cellular level might enhance BiP actions to improve ER quality control and overall cell health. But how that might decrease misfolding of what is mostly, if not entirely, cytosolic mutant SOD1 seems elusive. Nevertheless, Sil1 might be of benefit in these situations.
References:
Hadzipasic M, Tahvildari B, Nagy M, Bian M, Horwich AL, McCormick DA. Selective degeneration of a physiological subtype of spinal motor neuron in mice with SOD1-linked ALS. Proc Natl Acad Sci U S A. 2014 Nov 25;111(47):16883-8. Epub 2014 Nov 10 PubMed.
Bandyopadhyay U, Cotney J, Nagy M, Oh S, Leng J, Mahajan M, Mane S, Fenton WA, Noonan JP, Horwich AL. RNA-Seq profiling of spinal cord motor neurons from a presymptomatic SOD1 ALS mouse. PLoS One. 2013;8(1):e53575. PubMed.
View all comments by Arthur HorwichMake a Comment
To make a comment you must login or register.