First Evidence for Transmitted Alzheimer’s Disease?
Quick Links
Research over the last decade has shown that aggregated Aβ seeds can be transferred between people during rare medical procedures, sparking amyloidosis in the recipient. Could this result in full-blown Alzheimer’s disease? In the January 29 Nature Medicine online, researchers led by John Collinge at University College London offer the first evidence for this. Collinge runs the U.K.’s National Prion Clinic, which evaluates neurological disease in people who, as children, were treated with growth hormone or tissue taken from cadavers. Out of a total of eight people referred to this clinic since 2017, most of whom were diagnosed with dementia or cognitive problems in their mid-40s, four were positive for Alzheimer’s biomarkers. In one person who died with mild cognitive impairment, an autopsy found widespread amyloid plaques and neurofibrillary tangles in the cortex. All eight had received growth hormone preparations known to be contaminated with Aβ seeds.
- Several people who received Aβ-contaminated growth hormone in childhood developed dementia in midlife.
- Four of them had biomarker or pathological evidence of Alzheimer’s disease.
- This is the first evidence of iatrogenic AD transmission.
“[The authors] provide tantalizing evidence that, under extraordinary circumstances, Alzheimer’s disease is transmissible by a prion-like mechanism,” Mathias Jucker at the University of Tübingen, Germany, and Lary Walker at Emory University, Atlanta, noted in an accompanying editorial.
Experts agreed. “This is a very important and impactful study providing the first convincing evidence that AD can be transmitted in humans, and that the incubation time is around 35 years,” Jochen Herms at Ludwig-Maximilians University, Munich, wrote to Alzforum. David Knopman at the Mayo Clinic in Rochester, Minnesota, concurred, “These observations show iatrogenic transmission of Alzheimer pathology is possible.”
At the same time, they caution that the findings fall short of definitive proof of iatrogenic AD. “Pathological evidence, so far available in two of three deceased patients, suffered either from incomplete sampling or was too sparse to allow a state-of-the-art AD diagnosis,” said Herbert Budka at the Medical University in Vienna. Gaël Nicolas at Rouen University Hospital, France, noted that sporadic Alzheimer’s pathology can start in midlife. “Overall, I feel it difficult to be sure that AD-related changes were of iatrogenic origin,” he wrote (comments below).
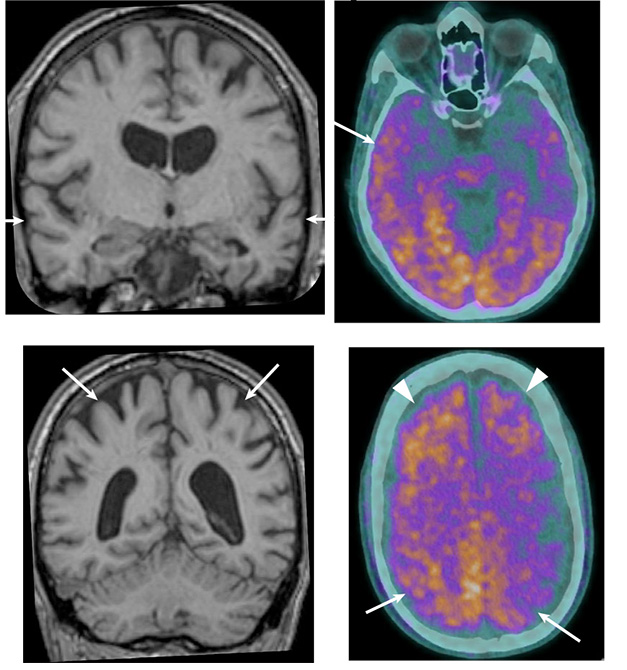
Alzheimer’s Evidence. A middle-aged man exposed to Aβ seeds in childhood had temporal lobe (top left) and parietal lobe (bottom left) atrophy (arrows), as well as amyloid PET tracer uptake in frontal, temporal, and parietal lobes (right). [Courtesy of Banerjee et al., Nature Medicine.]
A Mixed Clinical Picture
Previously, Collinge and colleagues at the NPC had found cerebral amyloid angiopathy (CAA) and parenchymal Aβ in seven out of eight people who died in their 40s from iatrogenic Creutzfeldt-Jakob Disease transmitted by contaminated growth hormone preparations (Sep 2015 news). The authors went on to show that archived vials of the suspect growth hormone extract contained Aβ seeds that triggered amyloidosis in mice, making this the likely cause of the early onset AD pathology (Dec 2018 news). Still, none of the CJD patients had Alzheimer’s symptoms, and their pathology did not meet the criteria for AD, leaving unclear whether they would have developed the disease had they lived longer.
In the new work, first author Gargi Banerjee and colleagues reported case studies from the next eight patients referred to the NPC, all of whom had received injections of the contaminated batches of growth hormone. At the time of referral, five of them had dementia; one had mild cognitive impairment, one, subjective cognitive impairment, and one was cognitively healthy. Two of them had memory problems characteristic of Alzheimer’s, while the others had varied deficits including language difficulties, problems with executive function, and behavior changes. Their symptoms began at a mean age of 46. Four of the eight had pathological or biomarker evidence consistent with AD.
The sole pathological evidence came from the man with MCI, who died at age 47 after a childhood brain tumor had returned. In his neocortex, the investigators found a moderate density of neuritic plaques, equivalent to CERAD stage 2, along with severe CAA. He had neurofibrillary tangles in his insular cortex, but because medial temporal lobe samples were unavailable, his Braak stage could not be determined. His cognitive changes were mostly behavioral.
Another man, who is still living, developed language and memory problems at age 38 and was diagnosed with early onset AD 10 years later. At that time, amyloid PET showed widespread plaques in his frontal, parietal, and temporal lobes, and his cerebrospinal fluid measures of Aβ42 and total tau met the cutoff for Alzheimer’s. MRI showed shrinkage in his hippocampus and temporal lobe (see image above).
Likewise, a woman who began having memory and language problems at age 46 was diagnosed with EOAD. She did not have fluid biomarker testing, but MRI revealed medial temporal lobe atrophy characteristic of AD. The fourth case was a 57-year-old man without cognitive symptoms, but with CSF Ab42/40 and p-tau181 in the AD range.
The other four cases lacked evidence specific for Alzheimer’s. One woman died at age 57 with dementia, but her autopsy showed only diffuse amyloid deposits, no plaques or tangles. Another woman died at 54 with dementia that primarily manifested via language and behavior problems. An autopsy was not done, but a previous MRI indicated frontal atrophy. A 48-year-old man with amnestic dementia initially had CSF Aβ42/40 that was borderline for AD, but repeat testing generated values in the normal range for Aβ42/40, p-tau181, and total tau. Finally, a 52-year-old man with subjective cognitive impairment was negative on amyloid PET and CSF biomarkers.
None of the eight patients had known AD genes, and their medical histories suggested no risk for dementia. Banerjee and colleagues believe the overall data imply that at least some of these patients acquired iatrogenic AD from growth hormone injections, despite atypical clinical symptoms. Since acquired prion diseases often present differently than sporadic forms, it would make sense that the same thing could happen in Alzheimer’s, they argued.
Some commenters found the data persuasive. “The authors … provide convincing arguments why the iatrogenic exposure to Aβ seeds seems to be the most likely underlying cause,” Barbara Stopschinski at the University of Texas Southwestern Medical Center in Dallas wrote to Alzforum (comment below).
Others wanted a more complete demonstration. “Definitive proof … must await neuropathological demonstration of the disease. None of the brains that underwent postmortem examination fulfilled currently accepted criteria for diagnosis of AD,” Seth Love at the University of Bristol, U.K., wrote to Alzforum (comment below).
What Does It Mean for Public Health?
Despite these differences of opinion, researchers agreed that the findings are not cause for alarm. Use of cadaveric brain tissue was once common in Europe and the U.S., but was discontinued in 1985. Around 1,800 people in the U.K. were treated with cadaver-derived growth hormone injections; of those, two-thirds are estimated to have received the contaminated preparation, Banerjee said in a press conference. Researchers have identified 80 cases of iatrogenic CJD in this group, but no AD until now. There may be fewer recognized AD cases because amyloidosis progresses more slowly than prion disease, the authors speculated.
In people exposed to Aβ seeds, vascular amyloidosis seems to be more common than AD. Researchers have now identified more than 100 cases of CAA in people who received dura mater grafts or had neurosurgery as children (Jan 2016 news; Feb 2018 news; Jan 2019 news). Steven Greenberg at Massachusetts General Hospital, Boston, noted that in all iatrogenic CAA cases that have come to autopsy, some sparse parenchymal deposits were present as well. “Identifying the factors that favor AD progression versus CAA progression remains an important area of study,” he wrote to Alzforum.
Although high-risk practices have been stopped, researchers are still grappling with the risk of potentially transmitting Aβ seeds during more common procedures such as neurosurgery or blood transfusions (Sep 2023). A white paper on this topic called for common-sense measures such as enzymatic cleaning of surgical instruments and using separate tools for children and adults (Sep 2020 news), while an NIH panel recommended more research into how aggregated proteins spread (Oct 2020 news).
Giovanna Lalli at the U.K. Dementia Research Institute in London, a co-first author on the white paper, said that these recommendations led the U.K. to tighten laboratory safety standards (Mead and Evans, 2021). Budka said the new findings should not change these practices. “Conclusions and recommendations in recent documents by international consortia still make sense,” he wrote (comments below).
Neurosurgeon Ville Leinonen at Kuopio University Hospital, Finland, believes the risk of accidental Aβ transmission during neurosurgery is quite low using current sterilization protocols, though he also recommends more study of the issue. Resources such as FinRegistry, which tracks lifetime surgical procedures for each patient, could help, he suggested (Viippola et al., 2023). Meanwhile, Colin Masters and Steven Collins at the University of Melbourne, Australia, suggested using low-cost fluid biomarkers to screen for signs of preclinical AD in people who had childhood procedures that may have put them at risk (comments below).
Could Aβ Treatments Foster Resistance?
With regard to understanding mechanisms of pathogenesis, researchers said the new findings strengthen the evidence for prion-like transmission of AD.
The authors suggest one potential consequence for therapies: If aggregated Aβ exists as a soup of different conformations in the brain, as do prion strains, then treatments that clear a specific form could allow minor species to take over, thus creating resistance to the medication. This has been described in cell culture with prion treatment (Oelschlegel and Weissmann, 2013; Bartz et al., 2021).
How likely is this? Some studies have found evidence of distinct Aβ strains in different subtypes of AD, but it is unclear if multiple conformations are present in a single brain (Jan 2017 news; Jan 2022 news).
Dieter Willbold of Heinrich-Heine University in Düsseldorf, Germany, thinks it is possible in principle for Aβ aggregates to develop resistance to therapeutic agents that have a strain-specific effect (comment below). Willbold has a small molecule in Phase 2 that binds Aβ monomers, preventing oligomer formation and stopping strains from forming (Nov 2023 news).
It is unknown if current immunotherapies are specific to a particular conformation. Most appear to bind Aβ across a range of sizes and forms, from oligomers to fibrils and mature plaques (e.g., Nov 2021 conference news).
Beyond AD, Banerjee et al.’s findings hint that other proteopathic diseases could have rare, acquired forms as well. “This work adds important data to support the idea that prion biology is not limited to prion protein, and probably extends to other proteins such as Aβ, tau, and α-synuclein,” Marc Diamond at UT Southwestern wrote to Alzforum (comment below). Jucker and Walker agree. “Given the growing list of disorders in which misfolded proteins are a defining feature, the expanded prion paradigm may well become one of the most important disease principles to have emerged in the past 50 years,” they wrote in their editorial.—Madolyn Bowman Rogers
References
News Citations
- Alzheimer’s Transmission Between People? Amyloid Plaques in Hormone Recipients Hint at Prion-like Spread
- Confirmed: Human Pituitary Extract Linked to Amyloidosis Contains Aβ Seeds
- News Brief: More Evidence for Aβ Spread Between People
- Can Aβ Seeds Be Transferred During Neurosurgery?
- First In Vivo Look at Amyloidosis Sparked by Dural Grafts
- Could Blood Transfusions Transmit Vascular Amyloid?
- Could Contaminated Scalpels Seed Amyloidosis?
- NIA Weighs in on Neurodegenerative Disease Transmissibility
- Do Palettes of Aβ Fibril Strains Differ Among Alzheimer’s Subtypes?
- Cryo-EM Unveils Distinct Aβ42 Fibril Structures for Sporadic, Familial AD
- After Long Wait, Aβ Oligomer Detangler Poised for Phase 2
- Lecanemab Sweeps Up Toxic Aβ Protofibrils, Catches Eyes of Trialists
Paper Citations
- Mead S, Evans T, Advisory Committee for Dangerous Pathogens Transmissible Spongiform Encephalopathy Subgroup. Safe laboratory management of prions and proteopathic seeds. Lancet Neurol. 2021 Dec;20(12):981. PubMed.
- Viippola E, Kuitunen S, Rodosthenous RS, Vabalas A, Hartonen T, Vartiainen P, Demmler J, Vuorinen AL, Liu A, Havulinna AS, Llorens V, Detrois KE, Wang F, Ferro M, Karvanen A, German J, Jukarainen S, Gracia-Tabuenca J, Hiekkalinna T, Koskelainen S, Kiiskinen T, Lahtela E, Lemmelä S, Paajanen T, Siirtola H, Reeve MP, Kristiansson K, Brunfeldt M, Aavikko M, Gen F, Perola M, Ganna A, FinnGen. Data Resource Profile: Nationwide registry data for high-throughput epidemiology and machine learning (FinRegistry). Int J Epidemiol. 2023 Aug 2;52(4):e195-e200. PubMed.
- Oelschlegel AM, Weissmann C. Acquisition of drug resistance and dependence by prions. PLoS Pathog. 2013 Feb;9(2):e1003158. Epub 2013 Feb 7 PubMed.
- Bartz JC. Environmental and host factors that contribute to prion strain evolution. Acta Neuropathol. 2021 Jul;142(1):5-16. Epub 2021 Apr 25 PubMed.
Further Reading
News
- As Parkinson’s Progresses, Do Synuclein Fibrils Shape-Shift?
- Does Fast-Progressing Alzheimer's Have a Whole Repertoire of Tau Conformers?
- Flock of New Folds Fills in Tauopathy Family Tree
- Traumatic Tau: Filaments from CTE Share Distinct Structure
- Conformers Confirmed: Structure of Pick’s Tau Distinct from AD Tau
Primary Papers
- Banerjee G, Farmer SF, Hyare H, Jaunmuktane Z, Mead S, Ryan NS, Schott JM, Werring DJ, Rudge P, Collinge J. Iatrogenic Alzheimer's disease in recipients of cadaveric pituitary-derived growth hormone. Nat Med. 2024 Feb;30(2):394-402. Epub 2024 Jan 29 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Ludwig-Maximilians-Universität Munich
This is a very important and impactful study providing first convincing evidence that AD can be transmitted in humans and that the incubation time is around 35 years.
I am missing a little the evaluation of the findings in a wider epidemiological context: e. g., what is the frequency of about 55-year-old AD patients (without family history and positive genetic testing) who did not receive c-hGH, and how does this compare to this cohort?
Although it seems likely that the transmission in the cases presented occurred via the application of contaminated c-hGH, these findings must also raise the important question of whether and how neurosurgical interventions in childhood per se (for example due to instruments contaminated with seeds which are not inactivated by standard procedures) may increase the risk for iatrogenic disease transmission, since four out of the eight patients of this study had undergone neurosurgery in childhood.
I miss a survey in this paper of whether AD occurs in younger age in case patients had a neurosurgical intervention about 35 years before the start of AD symptoms.
Mayo Clinic College of Medicine
The carefully presented observations by Banerjee et al. show that it is likely that Alzheimer pathology can be transmitted iatrogenically. From a clinical perspective, I hope that the lay press doesn’t over-react to this finding because the practical risk of iatrogenic transmission of Alzheimer pathology is zero. From a conceptual perspective, these observations are important in showing iatrogenic transmission of Alzheimer pathology is possible. While it is reasonable to assume, as did the authors, that Aβ species alone are the pathogenic molecule, these observations do not prove that claim.
Medical University Vienna
This work continues the clinical follow-up of a U.K. cohort of at least 1,848 individuals who had been treated during childhood with cadaver-derived growth hormone (Swerdlow et al., 2003). The cohort has emerged as an invaluable tool to allow a new glimpse on the etiology of neurodegeneration decades after the original treatment.
First, the tragedy of iatrogenic Creutzfeldt-Jakob disease (iCJD) by contamination of c-hGH with prion protein has struck 80 victims in this cohort (NCJDSU 2022). Second and more recently, demonstration of Aβ deposits mainly in vessel walls of iCJD brains of this cohort (Jaunmuktane et al., 2015)—caused by contamination with Aβ in the GH preparations (Purro et al., 2018) —or of patients who had undergone dural transplantations (Frontzek et al., 2016; Kovacs et al., 2016), culminated in a diagnostic framework for clinicians of iatrogenic congophilic angiopathy (iCAA, Banerjee et al., 2022). Importantly, in these cases of “iCAA,” brain parenchymal Aβ plaques were seen as well, whereas tau pathology was absent or not prominent.
Now third and most intriguingly, but not completely surprising, a detailed clinical study of eight further individuals with a spectrum of signs and symptoms three to four decades after c-hGH is presented. “The only factor common to all cases was the HWP subtype of c-hGH”–previously found to contain Aβ seeds in an experimental study (Purro et al., 2018). It was concluded to term the clinical syndrome therefore “iatrogenic AD, and AD should now be recognized as a potentially transmissible disorder."
But is this indeed AD? Not quite, at least as we have learnt to know it. First, Aβ is not AD. Second, a close look on the individual data reveals many difficulties inherent to clinical long-term follow up studies. Pathological evidence, so far available in two of three deceased patients, suffered either from incomplete sampling or was too sparse to allow a state-of–the-art AD diagnosis.
In fact, the illustrated Aβ deposits in brain parenchyma did not much differ from previous illustrations of parenchymal plaques in iCAA after c-hGH or dural grafting, whereas only one focal area in one brain also had prominent tau pathology, something next to a sulcus, reminiscent of the focal tau pathology seen in chronic traumatic encephalopathy.
ApoE and other AD risk genes were examined in five cases, with ε4 in one but without additional abnormalities. Clinical presentations were early onset dementia in five, MCI, subjective cognitive impairment, and lack of symptoms in one each. Two patients met AD biomarker data according to AT(N). Some presentations appeared atypical, and NIA-AA, DSM-V, or AT(N) criteria were variably met.
So, does this mean this report has little utility? In my view, no. Whatever name is given to this small series, its heterogeneity may be not only methodological. As rightly argued by the authors, phenotypical heterogeneity is notorious in neurodegenerative diseases such as sporadic, inherited and iatrogenic prion diseases, frontotemporal degenerations, extrapyramidal and motor neuron diseases, and AD as well. There, its molecular underpinning is only starting to be understood; distinct Aβ “strains” might cause deviation from classical clinico-pathological profiles.
While I do not think that the present paper definitively establishes “iatrogenic AD,” it is a first interesting and important step of the relevant scenario, starting with more complete clinico-pathological cases, as has happened successfully in the previous case of iCAA.
Importantly for public health risk assessment and management of potential transmission of Aβ, it must be emphasized that both proven transmission pathways, c-hGH as presented here and allografts of dura mater, have long since been discontinued. Neurosurgical procedures remain a possibility, although only few potential cases are on record (Jaunmuktane et al., 2018). In my view, the conclusions and recommendations in recent documents by international consortia still make sense (Lauwers et al., 2020; Asher et al., 2020).
References:
Swerdlow AJ, Higgins CD, Adlard P, Jones ME, Preece MA. Creutzfeldt-Jakob disease in United Kingdom patients treated with human pituitary growth hormone. Neurology. 2003 Sep 23;61(6):783-91. PubMed.
Jaunmuktane Z, Mead S, Ellis M, Wadsworth JD, Nicoll AJ, Kenny J, Launchbury F, Linehan J, Richard-Loendt A, Walker AS, Rudge P, Collinge J, Brandner S. Erratum: Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature. 2015 Oct 22;526(7574):595. Epub 2015 Sep 16 PubMed.
Purro SA, Farrow MA, Linehan J, Nazari T, Thomas DX, Chen Z, Mengel D, Saito T, Saido T, Rudge P, Brandner S, Walsh DM, Collinge J. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature. 2018 Dec;564(7736):415-419. Epub 2018 Dec 13 PubMed.
Frontzek K, Lutz MI, Aguzzi A, Kovacs GG, Budka H. Amyloid-β pathology and cerebral amyloid angiopathy are frequent in iatrogenic Creutzfeldt-Jakob disease after dural grafting. Swiss Med Wkly. 2016;146:w14287. Epub 2016 Jan 26 PubMed.
Kovacs GG, Lutz MI, Ricken G, Ströbel T, Höftberger R, Preusser M, Regelsberger G, Hönigschnabl S, Reiner A, Fischer P, Budka H, Hainfellner JA. Dura mater is a potential source of Aβ seeds. Acta Neuropathol. 2016 Jun;131(6):911-23. Epub 2016 Mar 25 PubMed.
Banerjee G, Samra K, Adams ME, Jaunmuktane Z, Parry-Jones AR, Grieve J, Toma AK, Farmer SF, Sylvester R, Houlden H, Rudge P, Mead S, Brandner S, Schott JM, Collinge J, Werring DJ. Iatrogenic cerebral amyloid angiopathy: an emerging clinical phenomenon. J Neurol Neurosurg Psychiatry. 2022 May 16; PubMed.
Jaunmuktane Z, Quaegebeur A, Taipa R, Viana-Baptista M, Barbosa R, Koriath C, Sciot R, Mead S, Brandner S. Evidence of amyloid-β cerebral amyloid angiopathy transmission through neurosurgery. Acta Neuropathol. 2018 May;135(5):671-679. Epub 2018 Feb 15 PubMed.
Lauwers E, Lalli G, Brandner S, Collinge J, Compernolle V, Duyckaerts C, Edgren G, Haïk S, Hardy J, Helmy A, Ivinson AJ, Jaunmuktane Z, Jucker M, Knight R, Lemmens R, Lin IC, Love S, Mead S, Perry VH, Pickett J, Poppy G, Radford SE, Rousseau F, Routledge C, Schiavo G, Schymkowitz J, Selkoe DJ, Smith C, Thal DR, Theys T, Tiberghien P, van den Burg P, Vandekerckhove P, Walton C, Zaaijer HL, Zetterberg H, De Strooper B. Potential human transmission of amyloid β pathology: surveillance and risks. Lancet Neurol. 2020 Oct;19(10):872-878. Epub 2020 Sep 16 PubMed.
Asher DM, Belay E, Bigio E, Brandner S, Brubaker SA, Caughey B, Clark B, Damon I, Diamond M, Freund M, Hyman BT, Jucker M, Keene CD, Lieberman AP, Mackiewicz M, Montine TJ, Morgello S, Phelps C, Safar J, Schneider JA, Schonberger LB, Sigurdson C, Silverberg N, Trojanowski JQ, Frosch MP. Risk of Transmissibility From Neurodegenerative Disease-Associated Proteins: Experimental Knowns and Unknowns. J Neuropathol Exp Neurol. 2020 Nov 1;79(11):1141-1146. PubMed.
Rouen University Hospital
This interesting report asks the critical important question, based on previous data in human and mice, of finding clinical cases of iatrogenic AD in cadaveric pituitary-derived growth hormone recipients beyond the reports of Aβ deposits in brains of individuals. Among the eight cases referred to the authors, I found striking the heterogeneity of phenotypes, including the unexpectedly high proportion of non-amnestic presentations. I wonder—as the authors discuss—whether all these cases actually have/had Alzheimer disease.
Based on the detailed case reports, I understand that three cases show clear evidence of AD-related changes at young ages, or even a diagnosis of early onset AD for two of them. Case 1 suffered from craniopharyngioma and also had EOAD confirmed at autopsy despite a non-amnestic presentation, without genetic data available; case 3 had EOAD with positive CSF biomarkers and a negative genetic screen, while case 6 is an asymptomatic person with CSF biomarkers consistent with presymptomatic AD, although he has normal cognition. I understand the latter is a research participant in a cohort. It would be interesting to know the CSF biomarkers of other research participants, and if he is the only case with such biological changes in the CSF, as well as how many individuals in this cohort had AD CSF biomarkers measured in total.
Among the other cases, two seem to have an intermediate level of evidence, as they presented dementia but without neuropathological examination nor CSF biomarkers available, so they could still exhibit another cause of dementia, related or no. Case 4 and case 5, the latter one presenting non-amnestic cognitive impairment with frontal atrophy, not allowing an AD diagnosis in absence of biomarkers available, which could even suggest FTD.
The other three cases are even more difficult. Case 2 cannot be diagnosed with AD, given the neuropathological examination, and despite diffuse Aβ deposits (but almost no neurofibrillary tangles and no plaques, Thal stage 1), with craniopharyngioma extending to the brain tissue, so maybe contributing to the symptoms. Still, diffuse Aβ deposits remain interesting in that case. Case 8 had dementia but normal AD CSF biomarkers (except 42/40 Aβ ratio considered borderline and then in normal ranges after a second lumbar puncture). It is unclear why case 7 was included here (subjective cognitive impairment with normal amyloid PET imaging and CSF AD biomarkers, thus arguing against AD), probably for the completeness of the report, i.e., reporting all eight referred cases?
There remains the question of what was the cause of dementia for the suspected non-AD cases, especially considering that some have important comorbidities. Even while some cases are not so clear, some clearly show biological changes in favor of an AD-related process or even confirmed or highly likely EOAD.
Overall, it seems that there are four of eight cases with evidence of amyloid pathology, but only two EOAD cases with dementia and supporting biomarkers or neuropathology, if I understood the clinical cases correctly. That they were all treated with the same preparation technique, and received a high level of ascertainment by this well-known institution, makes them very intriguing and the conclusions interesting, in light of prior evidence of the presence of seeds in the preparations and even transferability to mice.
I wonder what is the denominator here. How many individuals got these injections with same preparation types/batches overall at the scale of the country, and if prevalence of EOAD/amyloid changes would significantly vary from that in the general population? Absence of monogenic cause is not proof of iatrogenic cause, as more than 85 percent of EOAD cases do not have monogenic AD, and AD is common. We know that Aβ deposits are common in the population and do not always translate into AD in the lifetime, and that pathological changes leading to AD begin years or decades prior to symptom onset, so it is not surprising that some cadaveric preparations had Aβ seeds. Thus, if such a transmission is confirmed, we would expect many more cases than those of CJD, as the authors suggest. However, we don't know the role of the host response, and the variance of the incubation period. I think it will be difficult to show final evidence that some of the cases are actually iatrogenic, given the high frequency of AD, unless there is a clear increase in AD incidence in recipients, with statistical evidence that these individuals are actually more at risk of developing AD than in the general population. Easily said, but difficult to demonstrate.
Overall, I feel it difficult to be sure that AD-related changes were of iatrogenic origin in the cases with biological evidence of AD pathophysiology, and the other cases remain unexplained. I am looking forward to the follow-up of the cohort. The detailed description of these cases is an important added value to the literature.
UT Southwestern, Dallas
This is a very thorough report on a group of patients who received human cadaveric pituitary-derived growth hormone containing Aβ seeds in childhood for variable conditions and developed early onset of dementia with AD-like features in their 40s and 50s. The supplement of this paper contains highly informative case reports for each patient. It is interesting that in the majority of cases, the clinical trajectory was not typical for either inherited or sporadic AD; this suggests an atypical etiology. In some cases, additional biomarker data confirmed the presence of AD pathology based on ATN criteria.
The authors discuss possible etiologies of the clinical and pathological findings and provide convincing arguments why the iatrogenic exposure to Aβ seeds seems to be the most likely underlying cause. The authors conclude that AD is transmissible in certain exceptional situations such as infusion of pituitary extracts that contain Aβ seeds.
Taken together, this report extends prior work on the iatrogenic transmission of Aβ and provides additional evidence that AD could have the same etiologies that are known for conventional prion diseases, i.e., sporadic, inherited, and acquired/iatrogenic. This paper, in conjunction with previous reports on Aβ transmission, will be important to guide future investigations into the circumstances that could lead to Aβ transmission.
Based on preclinical and clinical evidence so far, Aβ transmission requires pooled seeds administered under exceptional circumstances; i.e., intravenous infusions, intracerebral injection. It is important to emphasize that there is no evidence for Aβ transmission in other contexts, for example, during routine surgeries or activities of daily life.
The authors also discuss a possible role for Aβ strains leading to distinct clinical phenotypes, and caution that new anti-Aβ drugs may select for dominant species and enhance propagation of non-dominant Aβ strains. More experimental and clinical data is needed to test the role of Aβ strains in humans, but if true, this could have important implications for ongoing and future treatment efforts. Besides strain subtype, other factors such as inflammation, genetic disposition, etc., could have contributed to the propagation of Aβ seeds and development of a dementia syndrome in this patient cohort and will require further investigation in future studies.
In summary, the paper adds compelling evidence that the principles of prion biology apply to Aβ and raises the question if these principles apply to other disease-associated amyloidogenic proteins such as tau and α-synuclein.
University of Bristol
This carefully considered, well-argued analysis of a small case series strongly suggests that iatrogenic transmission of Aβ in the course of human cadaver-derived pituitary growth hormone replacement therapy can induce not only CAA and diffuse Aβ plaques but also cerebral tau pathology and probably AD. That might not be surprising, given the established role of Aβ accumulation in the development of AD.
However, whilst the biochemical findings supported a diagnosis of AD in several of the patients, I think definitive proof of the development of iatrogenic AD in cadaver-derived pituitary growth hormone recipients must await neuropathological demonstration of the disease. None of the brains that underwent postmortem examination fulfilled currently accepted criteria for diagnosis of AD. Indeed, the pathological substrate of the dementia in the one patient who had definite dementia and whose brain was examined is not at all clear. It is noteworthy that that patient was one of those who had had radiosurgical treatment of his tumor, although the authors make strong arguments that that was probably not relevant to his cognitive decline. No mention is made of the postmortem neuropathological findings in the hypothalamus, which would have received most of the irradiation.
In reviewing the possible explanations for the development of cognitive impairment or dementia in cadaver-derived pituitary growth hormone recipients, the authors could perhaps have considered CAA itself, the one type of AD-associated cerebral pathology for which iatrogenic transmission has been most convincingly demonstrated. It is true that most previous cases of iatrogenic CAA have been identified as a result of their presenting with ICH. There will, however, certainly be other individuals with iatrogenic CAA who have not presented with ICH, and we know, at least in the context of sporadic disease, that CAA is a significant cause or contributor to dementia (see, for example, Keage et al., 2009).
The authors draw several practical conclusions from this case series. I would add one more—that more postmortem neuropathological studies are needed in individuals who in life were exposed to possible iatrogenic transmission of Aβ.
References:
Keage HA, Carare RO, Friedland RP, Ince PG, Love S, Nicoll JA, Wharton SB, Weller RO, Brayne C. Population studies of sporadic cerebral amyloid angiopathy and dementia: a systematic review. BMC Neurol. 2009 Jan 13;9:3. PubMed.
UK Dementia Research Institute
This very interesting paper is, as far as I know, the first demonstration of potential iatrogenic AD; previous cases had pointed to iatrogenic CAA. As cases are extremely rare, I think more research is warranted. The findings are not entirely surprising given previous literature (reviewed in Lauwers et al., 2020).
Based on current evidence, the risk of acquiring a transmissible form of amyloid is very low. No one should reconsider or forego any medical procedure, especially for blood transfusion or neurosurgery, which saves many lives worldwide every year. However, it is always important that we continue to review and scrutinize evidence where public health is concerned. We have previously called for increased vigilance and long-term monitoring, particularly following procedures in early life that involve human fluids or tissues. Practical steps recommended include conducting larger epidemiological studies, the development of low-cost, high-throughput sensitive tests for Aβ and other proteins to facilitate the precautionary sterilization of, for example, neurosurgical instruments.
In Lauwers et al., we offered a number of recommendations including the adoption of cautionary measures for the laboratory manipulation of purified proteopathic seeds. This subsequently led to the modification of safety guidance in the U.K. (Mead and Evans, 2021).
References:
Lauwers E, Lalli G, Brandner S, Collinge J, Compernolle V, Duyckaerts C, Edgren G, Haïk S, Hardy J, Helmy A, Ivinson AJ, Jaunmuktane Z, Jucker M, Knight R, Lemmens R, Lin IC, Love S, Mead S, Perry VH, Pickett J, Poppy G, Radford SE, Rousseau F, Routledge C, Schiavo G, Schymkowitz J, Selkoe DJ, Smith C, Thal DR, Theys T, Tiberghien P, van den Burg P, Vandekerckhove P, Walton C, Zaaijer HL, Zetterberg H, De Strooper B. Potential human transmission of amyloid β pathology: surveillance and risks. Lancet Neurol. 2020 Oct;19(10):872-878. Epub 2020 Sep 16 PubMed.
Mead S, Evans T, Advisory Committee for Dangerous Pathogens Transmissible Spongiform Encephalopathy Subgroup. Safe laboratory management of prions and proteopathic seeds. Lancet Neurol. 2021 Dec;20(12):981. PubMed.
University of Eastern Finland
The study is extremely interesting on the hot topic of potential transmission of Aβ in similar fashion as prion protein. The advent of acquired “mad cow” disease led to new standards of instrument handling when CJD is suspected. This has arguably led to some degree of overkill in the expansion of disposable surgical instruments, which, sadly, goes counter to the general need for sustainable development.
I consider the risk of transmission of Aβ during neurosurgical procedures to be extremely small, or practically nonexistent, when the current standards of instrument sterilization processes are properly applied. However, high-quality research is urgently needed to verify or exclude the proposed potential risk of Aβ protein transmission. Along with experimental studies, the FinRegistry’s inclusion of time-stamped diagnoses and surgical procedures over the entire lifespan is an example of a highly valuable future tool for investigating such potential associations from an epidemiological perspective (Viippola et al. 2023).
References:
Viippola E, Kuitunen S, Rodosthenous RS, Vabalas A, Hartonen T, Vartiainen P, Demmler J, Vuorinen AL, Liu A, Havulinna AS, Llorens V, Detrois KE, Wang F, Ferro M, Karvanen A, German J, Jukarainen S, Gracia-Tabuenca J, Hiekkalinna T, Koskelainen S, Kiiskinen T, Lahtela E, Lemmelä S, Paajanen T, Siirtola H, Reeve MP, Kristiansson K, Brunfeldt M, Aavikko M, Gen F, Perola M, Ganna A, FinnGen. Data Resource Profile: Nationwide registry data for high-throughput epidemiology and machine learning (FinRegistry). Int J Epidemiol. 2023 Aug 2;52(4):e195-e200. PubMed.
University of Melbourne
University of Melbourne
Iatrogenic Alzheimer’s disease: a medical misadventure?
Premised on our current understanding of the continuum of pathogenic changes in AD evolution, the U.K. National Prion Monitoring Cohort (NPMC) now reports eight cases of likely young-onset (38-55 years old) Alzheimer’s disease (AD) from a cohort of more than 1,848 subjects who received cadaveric pituitary-derived human growth hormone (c-hGH) 30-44 years before onset. The cases include presymptomatic biomarker-supported subjects and those with amnestic and non-amnestic cognitive impairment with varying investigational and pathological corroboration. This report comes on a background of 80 cases of iatrogenic Creutzfeldt-Jakob disease (CJD) identified from the same cohort.
Separately illustrating inadvertent transmission, cases of overt cerebral Aβ angiopathy (CAA) are being increasingly recognized in 40- to 50-year-old subjects who received dura mater grafts or had other neurosurgical procedures as children. More than 100 such subjects have now been identified world-wide (Banerjee et al., 2022; Lauwers et al., 2020). All cases of pathologically confirmed AD have CAA, albeit to varying degrees. Those that present with overt vascular complications (hemorrhage, vasculitis) are a minority (probably less than 1 percent) of all AD subjects.
More than 50 years ago, the first evidence emerged that experimental forms of systemic (AA) amyloidosis might be transmissible (Lundmark et al., 2002) after it was determined that the amyloid-enhancing factor (AEF) was the serum amyloid A (SAA) protein itself. Around the same time, kuru and CJD were shown to be transmissible, like ovine scrapie, and finally a coherent concept of auto-catalytic infectious/transmissible amyloids was developed. Despite several false starts and repeated attempts, AD proved not to be easily transmissible, although Aβ showed some properties that, under certain conditions, enabled it to propagate after inoculation.
We are now faced with a serious public health issue where large numbers of people may be at risk of developing early onset AD/CAA because of childhood exposures through Aβ-contaminated neurosurgical instruments and implants, or treatments with human cadaveric pituitary-derived hormones (growth hormones and gonadotrophins).
Fortunately, we are now positioned to evaluate this risk through the use of biofluid (CSF, plasma) biomarkers of Aβ42 and p-tau, which are proving to be reliable in the detection of preclinical AD (Ashton et al., 2024). It will be cost-effective to screen for the development of preclinical AD in this at-risk population (with appropriate controls) and determine the extent of this risk. It is even more fortunate that the advent of disease-modifying immunotherapies targeting Aβ for symptomatic AD may have arrived just in time to be deployed in secondary prevention strategies, which are now being evaluated in clinical trials.
References:
Ashton NJ, Brum WS, Di Molfetta G, Benedet AL, Arslan B, Jonaitis E, Langhough RE, Cody K, Wilson R, Carlsson CM, Vanmechelen E, Montoliu-Gaya L, Lantero-Rodriguez J, Rahmouni N, Tissot C, Stevenson J, Servaes S, Therriault J, Pascoal T, Lleó A, Alcolea D, Fortea J, Rosa-Neto P, Johnson S, Jeromin A, Blennow K, Zetterberg H. Diagnostic Accuracy of a Plasma Phosphorylated Tau 217 Immunoassay for Alzheimer Disease Pathology. JAMA Neurol. 2024 Mar 1;81(3):255-263. PubMed.
Banerjee G, Samra K, Adams ME, Jaunmuktane Z, Parry-Jones AR, Grieve J, Toma AK, Farmer SF, Sylvester R, Houlden H, Rudge P, Mead S, Brandner S, Schott JM, Collinge J, Werring DJ. Iatrogenic cerebral amyloid angiopathy: an emerging clinical phenomenon. J Neurol Neurosurg Psychiatry. 2022 May 16; PubMed.
Lauwers E, Lalli G, Brandner S, Collinge J, Compernolle V, Duyckaerts C, Edgren G, Haïk S, Hardy J, Helmy A, Ivinson AJ, Jaunmuktane Z, Jucker M, Knight R, Lemmens R, Lin IC, Love S, Mead S, Perry VH, Pickett J, Poppy G, Radford SE, Rousseau F, Routledge C, Schiavo G, Schymkowitz J, Selkoe DJ, Smith C, Thal DR, Theys T, Tiberghien P, van den Burg P, Vandekerckhove P, Walton C, Zaaijer HL, Zetterberg H, De Strooper B. Potential human transmission of amyloid β pathology: surveillance and risks. Lancet Neurol. 2020 Oct;19(10):872-878. Epub 2020 Sep 16 PubMed.
Lundmark K, Westermark GT, Nyström S, Murphy CL, Solomon A, Westermark P. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc Natl Acad Sci U S A. 2002 May 14;99(10):6979-84. PubMed.
Heinrich Heine University Düsseldorf; Forschungszentrum Jülich; and Priavoid GmbH, Düsseldorf, Germany
If self-replicating protein aggregates are the trigger for Alzheimer's disease, then the disease must in principle be transmissible. It is as simple as that. After the transmissibility of the pathology has been demonstrated in animals and humans, the work of John Collinge and colleagues now shows that the transmitted pathology also induces the corresponding clinical symptoms and thus transmits the disease. This is a further recommendation of Aβ aggregates as a target for the treatment of Alzheimer's disease.
The possibility that there are several different strains of Aβ seeds makes it possible, conceptually, to develop resistance to therapeutic agents that have a strain-specific effect. The anti-prionic mechanism of action developed by us, which stabilizes monomers and thus prevents and even reverses aggregation (Kass et al., 2022), is independent of the respective strain, because the monomer is stabilized in its IDP conformation by the compound PRI-002 (alias RD2 in publications) developed by us. PRI-002 thus acts independently of the Aβ strain. A Phase 2 study with PRI-002 started a few weeks ago.
References:
Kass B, Schemmert S, Zafiu C, Pils M, Bannach O, Kutzsche J, Bujnicki T, Willbold D. Aβ oligomer concentration in mouse and human brain and its drug-induced reduction ex vivo. Cell Rep Med. 2022 May 17;3(5):100630. PubMed.
University of Texas, Southwestern Medical Center
The Collinge group has performed a measured, thoughtful, and careful report of dementia following iatrogenic exposure of people to pituitary extracts that contained Aβ seeding activity. The story is compelling to the extent that the group of patients were exposed to extracts with seeding activity, and developed an atypical (as far as we know), early onset dementia that has features of Alzheimer’s.
What is left uncertain from the article is the odds that such patients could have developed these disorders sporadically. I don’t know the answer to this, but it seems unlikely that their common exposure to pituitary extract was a random event. Furthermore, because of previous work by Mathias Jucker and Lary Walker, as well as others, to suggest transmissibility of Aβ pathology, this report is even more compelling.
Importantly, as the authors emphasize, absent extraordinary exposure (intravenous administration of pooled pituitary extracts), the risk of transmissibility of Aβ (or likely tau) to humans based on casual contact is essential nil.
Taken together, this work adds important data to support the idea that prion biology is not limited to PrP, and probably extends to other proteins such as Aβ, tau, and α-synuclein, among other amyloid proteins.
Karolinska Institutet
As far as I know, this is the first evidence supporting that Alzheimer’s (and not just CAA) is transmissible in humans. If Alzheimer’s were to be transmissible, that would certainly open up a range of questions and concerns, including whether other, more common medical procedures, such as blood transfusion or transplantations, could be transmission routes. However, given the long incubation times, I don’t think we are set up to detect these kinds of iatrogenic transmissions on a large scale, and so we would need to find smart ways of trying to assess that.
Henan Academy of Innovations in Medical Science
I would posit that the observations made can be explained by aging processes. If these patients received growth hormone in their childhood, one can assume that they are at risk for developmental abnormalities that result in abnormal brain aging. To claim that these people developed an AD-like state because they were exposed to microscopic amounts of amyloid when they were children, and 30-40 years later they develop a nearly AD like status, is a stretch.
We all have amyloid in our brains. The donors of growth hormone did not have AD, therefore it is unlikely that they had some form of unique oligomer that acts as a catalyst to induce an AD-like state in adults. This story needs a scientific model of how tiny amounts of amyloid could possibly induce AD decades later. People get blood transfusions all the time, some of which probably contain amyloid, and there is no AD epidemic coming from that.
In a commendable effort to prevent panic and shunning of AD patients, several expert commentators have taken pains to assert evidence so far shows minimal, if any, risk for transmission. However, the truth is that nobody knows. Several classic infections can transmit a "carrier state" without symptoms at once or ever. Host genetics also influence resistance to transmission.
Occupationally, neurosurgeons would seem most at risk for contact with an infectious agent. Indeed, a report on cause of death among these specialists surprised the authors by showing a higher number of AD cases than expected (Lollis et al., 2010). This group is at less risk now, as precautions mandated for HIV and hepatitis have inspired careful protection against contact with a patient's blood.
In the research lab, specimens of brain tissue and amyloid are often handled in cavalier fashion, with the researcher not wearing gloves or goggles. Yet, a wisp of evidence that some agent might be transferred came from George Glenner, pioneer in elucidation of brain amyloid, who died in 1995 of "systemic senile amyloidosis" (obituary, The New York Times).
While we await more evidence of transmission and its mechanism, it would seem sensible to attend carefully to precautionary measures in the operating room and research lab.
References:
Lollis SS, Valdes PA, Li Z, Ball PA, Roberts DW. Cause-specific mortality among neurosurgeons. J Neurosurg. 2010 Sep;113(3):474-8. PubMed.
CNRS, CEA, Molecular Imaging Research Center
Several studies have already shown that Aβ and tau pathologies are transmitted after iatrogeniuc contamination with Aβ/tau-contaminated compounds (Lauwers et al., 2020). The functional impact of such transmission was, however, still unclear as most subjects did not display AD-like clinical signs.
In two studies in primates, using two different cohorts, cognitive impairments as well as amyloid plaques and tau pathology were induced by intracerebral inoculation of human brain extracts presenting with both Aβ and tau (Gary et al., 2019; Lam et al., 2021). As the doses of Aβ and tau inoculated in primates was high, it remained unknown whether transmission of clinical signs could occur in humans following intramuscular or subcutaneous administration of Aβ/tau-contaminated c-hGH. The article by Banerjee et al. is thus critical as it suggests that symptomatic AD-like cognitive impairments can be transmitted by Aβ/tau-contaminated compounds. This novel form of AD is called iatrogenic AD by the authors.
However, the AD biomarker and postmortem results are intriguing. Postmortem tau deposits were sparse. This suggests that tau was not strongly transmitted and that Aβ did not strongly induce tau pathology in iatrogenic AD. It is widely acknowledged that tau pathology spreading is strongly associated with clinical signs in AD (Braak and Braak, 1991). In primates, tau also tends to be associated with cognitive impairments (Gary et al., 2019). Here, tau pathology obviously cannot explain the clinical signs. Aβ deposition can be the culprit in some cases (i.e., case 1) but not in other cases (e.g., case 2). The main culprits for the reported cognitive impairments will have to be identified in future studies.
References:
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239-59. PubMed.
Gary C, Lam S, Hérard AS, Koch JE, Petit F, Gipchtein P, Sawiak SJ, Caillierez R, Eddarkaoui S, Colin M, Aujard F, Deslys JP, French Neuropathology Network, Brouillet E, Buée L, Comoy EE, Pifferi F, Picq JL, Dhenain M. Encephalopathy induced by Alzheimer brain inoculation in a non-human primate. Acta Neuropathol Commun. 2019 Sep 4;7(1):126. PubMed.
Lam S, Petit F, Hérard AS, Boluda S, Eddarkaoui S, Guillermier M, Brain Bank Neuro-C. E. B. Neuropathology Network, Buée L, Duyckaerts C, Haïk S, Picq JL, Dhenain M. Transmission of amyloid-beta and tau pathologies is associated with cognitive impairments in a primate. Acta Neuropathol Commun. 2021 Oct 12;9(1):165. PubMed.
Lauwers E, Lalli G, Brandner S, Collinge J, Compernolle V, Duyckaerts C, Edgren G, Haïk S, Hardy J, Helmy A, Ivinson AJ, Jaunmuktane Z, Jucker M, Knight R, Lemmens R, Lin IC, Love S, Mead S, Perry VH, Pickett J, Poppy G, Radford SE, Rousseau F, Routledge C, Schiavo G, Schymkowitz J, Selkoe DJ, Smith C, Thal DR, Theys T, Tiberghien P, van den Burg P, Vandekerckhove P, Walton C, Zaaijer HL, Zetterberg H, De Strooper B. Potential human transmission of amyloid β pathology: surveillance and risks. Lancet Neurol. 2020 Oct;19(10):872-878. Epub 2020 Sep 16 PubMed.
Since the misfolded proteins have been found in eye tissues and tears—have medical procedures involving the eye been considered as possible transmission routes?
Might we consider that, if prions can induce Aβ seeding, microbes such as herpes simplex virus (HSV) and herpes varicella-zoster, which are transmissible, might as well? For example, studies have shown that HSV, other viruses, and many other types of organisms reside in the elderly brain, that HSV encephalitis damages localized regions in the limbic system related to memory and learning, and that rapid development of plaques and tangles in mice infected with HSV or bacteria supports this possibility (Itzhaki et al., 2016).
Viruses replicate intracellularly and spread from cell to cell. Untreated syphilis (Treponema pallidum) is a well-known cause of dementia, and a study of Borrelia burgdorferi, another spirochete that causes Lyme disease, showed that Borrelia-positive aggregates co-localized with amyloid and phospho-tau markers (Senejani et al., 2022). Other studies have demonstrated that Aβ is antimicrobial (Soscia et al., 2010), and epidemiological studies have reported that people who received shingles vaccines were significantly less likely to develop dementia (Lophatananon et al., 2021). Such infections may be preventable or even treatable triggers or accelerators of Alzheimer's and other neurological disorders and should be considered when evaluating people with symptoms of memory or other neurological impairments.
References:
Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H, Bullido MJ, Carter C, Clerici M, Cosby SL, Del Tredici K, Field H, Fulop T, Grassi C, Griffin WS, Haas J, Hudson AP, Kamer AR, Kell DB, Licastro F, Letenneur L, Lövheim H, Mancuso R, Miklossy J, Otth C, Palamara AT, Perry G, Preston C, Pretorius E, Strandberg T, Tabet N, Taylor-Robinson SD, Whittum-Hudson JA. Microbes and Alzheimer's Disease. J Alzheimers Dis. 2016;51(4):979-84. PubMed.
Senejani AG, Maghsoudlou J, El-Zohiry D, Gaur G, Wawrzeniak K, Caravaglia C, Khatri VA, MacDonald A, Sapi E. Borrelia burgdorferi Co-Localizing with Amyloid Markers in Alzheimer's Disease Brain Tissues. J Alzheimers Dis. 2022;85(2):889-903. PubMed.
Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010 Mar 3;5(3):e9505. PubMed.
Lophatananon A, Mekli K, Cant R, Burns A, Dobson C, Itzhaki R, Muir K. Shingles, Zostavax vaccination and risk of developing dementia: a nested case-control study-results from the UK Biobank cohort. BMJ Open. 2021 Oct 8;11(10):e045871. PubMed.
View all comments by Mary NewportMake a Comment
To make a comment you must login or register.