A First for Proteinopathies—Heteromeric Fibrils
Quick Links
In recent years, cryo-electron microscopists have unveiled a dazzling array of amyloid filament structures that underlie neurodegenerative diseases. Until now, all of them included but a single protein—Aβ, tau, α-synuclein, TDP-43, and others. That’s why Benjamin Ryskeldi-Falcon and colleagues at the MRC Laboratory of Molecular Biology in Cambridge, U.K., were shocked to find that in one type of frontotemporal lobar degeneration, two proteins join forces to form a single fibril. As reported in a preprint posted on bioRxiv on June 26, TDP-43 buddied up with Annexin A11 to form filaments with a kite-shaped core. This pairing was only found among cases of FTLD-TDP Type C, one of the most common forms of sporadic FTD with TDP-43 pathology. The kite is distinct from the TDP-43 contortions resolved for other neuropathological types of FTLD-TDP.
- Segments from TDP-43 and Annexin A11 snap together in a kite-shaped fold.
- An N-terminal fragment of Annexin A11 predominates in the fibrils.
- The proteins co-mingle in dystrophic neurites in FTLD-TDP Type C, but rarely in Types A or B.
These are the first heteromeric amyloid fibrils ever discovered in the human brain. The surprising structure also casts Annexin A11 (ANXA11)—a protein that helps shuttle lysosomes, RNA, and TDP-43 along axons—as a co-instigator in FTLD.
“By demonstrating the coherent integration of two different proteins into individual amyloid filaments, [the authors] convincingly revise the ‘one filament, one protein’ concept, and raise intriguing questions about amyloidogenesis in general,” Lary Walker of Emory University in Atlanta wrote to Alzforum.
Ian Mackenzie of the University of British Columbia thinks the findings have broad implications. “The identification of the first heteromeric filaments associated with neurodegenerative disease suggests a new way in which pathological proteins might interact that may be relevant to a number of other conditions,” he wrote (comments below).
The discovery coincides with a neuropathological analysis published June 19 in Acta Neuropathologica. It spied the same two culprits—TDP-43 and ANXA11—co-mingling in dozens of FTLD-TDP Type C brain samples. Scientists led by Sílvia Porta, Vivianna van Deerlin, and Edward Lee of the University of Pennsylvania in Philadelphia, also identified a person with a coding variant in ANXA11 and aggregates of the protein in their brain, sans TDP-43 fibrils.
Based on how TDP-43 accumulates within cells and distributes throughout the brain, FTLD-TDP-43 proteinopathies are classified into four types: A-D (Neumann et al., 2021). Previously, Ryskeldi-Falcon’s group resolved a double spiral-shaped fold at the core of filaments from people with FTLD-TDP Type B. It is typically found among people with a combination of FTD and ALS (Dec 2021 news). Later, the same researchers spotted a wholly different, chevron-shaped configuration in FTLD-TDP Type A, a form that most commonly underlies cases of behavioral variant FTD without ALS (Aug 2023 news).
This time, they honed in on filaments of Type C, which most commonly lurk in people with semantic variant primary progressive aphasia (svPPA), a sporadic form of FTD. In this dementia, elongated inclusions of the RNA-binding protein clog dystrophic neurites in the superficial layers of the cortex. These contrasts the inclusions that mostly haunt neuronal cell bodies in the other FTLD types.
To identify these neurite fibrils, first author Diana Arseni and colleagues extracted insoluble filaments from the prefrontal and temporal cortices of four people. From nearly 300,000 cryo-EM images, a single, left-twisting, TDP-43 filament structure emerged. Its core folded into a deltoid, comprising two separate chains of amino acids. At 2.7Ǻ resolution, one of these chains was clearly the low-complexity domain of TDP-43. The other was a mystery, until the researchers searched its primary sequence in reference proteomes. ANXA11 came back as the only hit. Strikingly, among the thousands of cryo-EM images they sifted through, the scientists did not find a single pure filament of TDP-43 or of ANXA11.
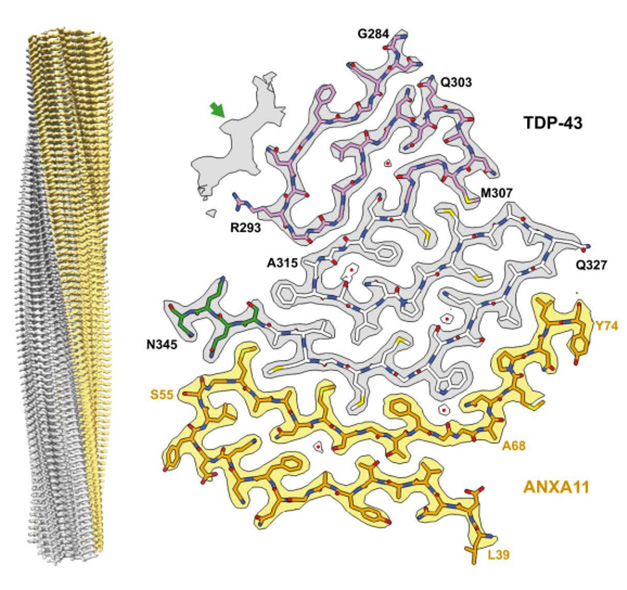
Double Trouble. A heteromeric, left-twisting filament (left) was found in all four people with FTLD-TDP Type C. Its core comprised segments of TDP-43 (gray) and Annexin A11 (yellow), snapped together in a deltoid configuration. [Courtesy of Arseni et al., bioRxiv, 2024.]
The protofilament core consisted of segments from each protein’s low-complexity domain. For TDP-43, this started with either G282 in an alternate configuration, or G284 in the predominant one, and spanned to N345. ANXA11 contributed residues L39-L74. The pair locked together via an extensive interface made of two antiparallel, largely hydrophobic β-sheets, with a kink in the center angling toward the ANXA11 side (image above). The remaining residues of ANXA11 folded back against this interface, forming another layer that extended to the central kink.
In serpentine style, TDP-43 coiled into four additional layers beyond the interface. Hydrophobic residues formed the first 1½ of these additional layers, while a glycine-rich segment rounded out the second layer and snaked into the third and fourth. Moving out from the interface, each layer was progressively shorter than the last, giving the core a triangular shape. In an alternate conformation, the glycine-rich region formed a single, pleated layer in place of the last two.
In classic amyloid filament style, stacks of these protofilaments were stabilized along the helical axis by hydrogen bonds between intermolecular, parallel β-sheets, by glutamine/asparagine side chain interactions, and by staggered hook-ups between aromatic side chains (image below).
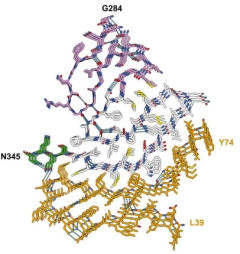
Serpentine Stack. An atomic model shows three layers of TDP-43/ANXA11 protofilaments. Hydrogen bonds (dashed cyan lines) between the protofilaments stabilize the structure. [Courtesy of Arseni et al., bioRxiv, 2024.]
Probing the isolated filaments via immunoblot, the researchers found that both full-length and C-terminal fragments of TDP-43 were able to contribute to the heteromeric filaments. For ANXA11, a 22kDa N-terminal fragment was predominant, while the full-length protein showed up in about 10 percent of filaments. How truncation of ANXA11 relates to the fibrils, and whether it happens before or after fibrils form, remains to be addressed, Ryskeldi-Falcon said. This N-terminal fragment was not detected in FTLD-TDP Type A or B.
Immunohistochemistry of prefrontal cortex samples gave credence to the specificity of the TDP-43 and ANXA11 relationship. Both proteins mingled within inclusions in dystrophic neurites and in neuronal cytoplasmic inclusions in Type C, but not Type A or B FTLD. In Type C, nary an inclusion contained one protein without the other.
The new TDP-43/ANXA11 hybrid structure joins a growing flock of TDP-43 filament folds resolved by Ryskeldi-Falcon’s group (image below). Next up will be filaments from FTLD-TDP Type D, a pathological pattern found exclusively in people with familial mutations in the VCP gene, Ryskeldi-Falcon told Alzforum.
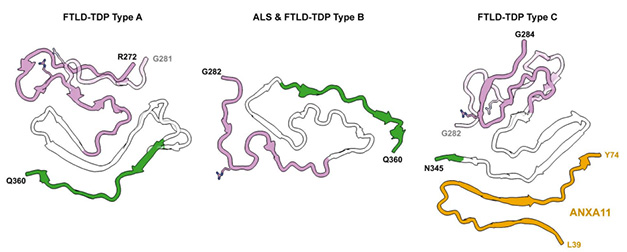
Twists of TDP-43. Previously identified TDP-43 filaments from Type A and Type B FTLD fold into chevron and double spiral configurations, respectively. Type C fibrils fold into a deltoid shape with ANXA11 (right). TDP-43 glycine-rich (pink), hydrophobic (white), and Q/N-rich (green) regions are designated. Alternate conformations of the glycine regions are transparent. [Courtesy of Arseni et al., bioRxiv, 2024.]
Neuropathological Alignment
For the most part, the new heteromeric structure identified in FTLD-TDP Type C cases complements results from Lee’s neuropathological study. Initially, co-first authors John Robinson and EunRan Suh and colleagues set out to hunt for rare genetic variants in ANXA11, and to study their neuropathological consequences. They identified five hits among 822 brain bank samples from the Center for Neurodegenerative Disease Research cohort at UPenn, including the previously described pathogenic variant, p.G38R, plus four of uncertain significance. Immunohistochemistry revealed ANXA11 inclusions in the pathogenic mutation sample, who had ALS, as well as in two of the other four. One of these had FTLD-TDP Type C, which is typically idiopathic.
This spurred Robinson to look for ANXA11 inclusions among 34 other FTLD-TDP Type C samples in the cohort. Lo and behold, all had them. These ANXA11 inclusions followed the pattern typical of TDP-43 aggregates in Type C pathology, for example by packing dystrophic neurites. The severity of ANXA11 and TDP-43 pathology tightly correlated across brain regions (image below). Dual immunofluorescence confirmed the co-localization of both proteins in inclusions. Notably, aggregated ANXA11 was truncated. Lee told Alzforum that this fragment is likely the same one that predominated in the heteromeric filaments Ryskeldi-Falcon found.

Partners in Pathology. In FTLD-TDP Type C, ANXA11 inclusions crowded dystrophic neurites in the temporal cortex (top left) and the neuronal cytoplasm in the hippocampus (top right). Severity of ANXA11 and TDP-43 pathology correlated tightly by brain region (bottom). [Courtesy of Robinson et al., Acta Neuropathologica, 2024.]
Outside of Type C, ANXA11 pathology was rare. That said, it did crop up in eight AD cases with limbic predominant TDP-43 encephalopathy (LATE-NC), two FTLD-TDP Type A cases, one person with FTLD-TDP Type B, and two people with ALS. These samples tended to have a high burden of mixed proteinopathies, and in each case, the pattern of “annexinopathy” followed that of TDP-43. Incidentally, a recent paper tied yet another a P93S ANXA11 variant to corticobasal degeneration (Snyder et al., 2024).
Lee acknowledged that for these non-Type C cases, it is unclear if the protein forms heteromeric filaments with TDP-43, or merely aggregates within the same inclusions.
Across the entire autopsy cohort, Robinson found but one example of annexinopathy without TDP-43 pathology, in a P75S carrier who had died with a disorder resembling progressive supranuclear palsy. The striatum had severely degenerated, while the putamen was ravaged by vacuoles like those seen in prion-like diseases. Neuritic inclusions of ANXA11 littered most brain regions except the cerebellum. The findings raise the possibility that ANXA11 pathology is sufficient to cause neurodegeneration independent of TDP-43 proteinopathy, the authors wrote. Lee said it would be interesting to determine the structure of any ANXA11 filaments in this case.
How is it that ANXA11 and TDP-43 team up only in FTLD-TDP Type C? The authors of both papers pointed out that, by virtue of their low-complexity domains, the two proteins are capable of undergoing liquid-liquid phase separation, thus gelling into condensed granules. Specifically, both are part of RNA-protein granules. Previously, Ward reported that ANXA11 tethers these granules to lysosomes, which are transported along axons to nerve terminals (Apr 2024 news). “One wonders if the residues bridging ANXA11 and TDP-43 within filamentous assemblies could functionally link these two proteins together within physiological RNPs, but also predispose them to pathological interactions during disease,” he wrote. Ryskeldi-Falcon noted that withering axons are unique to FTLD Type C, placing these two culprits right at the scene of the pathological crime.—Jessica Shugart
References
News Citations
- Double Spiral Sets TDP-43 Apart from Other Amyloids
- In Frontotemporal Lobar Dementia, TDP-43 Snaps into a Chevron Shape
- Axons or Bust: To Reach Neuron Extremities, RNAs Hitch a Ride on Lysosomes
Paper Citations
- Neumann M, Lee EB, Mackenzie IR. Frontotemporal Lobar Degeneration TDP-43-Immunoreactive Pathological Subtypes: Clinical and Mechanistic Significance. Adv Exp Med Biol. 2021;1281:201-217. PubMed.
- Snyder A, Ryan VH, Hawrot J, Lawton S, Ramos DM, Qi YA, Johnson KR, Reed X, Johnson NL, Kollasch AW, Duffy MF, VandeVrede L, Cochran JN, Miller BL, Toro C, Bielekova B, Marks DS, Yokoyama JS, Kwan JY, Cookson MR, Ward ME. An ANXA11 P93S variant dysregulates TDP-43 and causes corticobasal syndrome. Alzheimers Dement. 2024 Aug;20(8):5220-5235. Epub 2024 Jun 26 PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Arseni D, Nonaka T, Jacobsen MH, Murzin AG, Cracco L, Peak-Chew SY, Garringer HJ, Kawakami I, Suzuki H, Onaya M, Saito Y, Muryama S, Geula C, Vidal R, Newell KL, Mesulam M, Ghetti B, Hasegawa M, Ryskeldi-Falcon B. Heteromeric amyloid filaments of ANXA11 and TDP-43 in FTLD-TDP Type C. 2024 Jun 26 10.1101/2024.06.25.600403 (version 1) bioRxiv.
- Robinson JL, Suh E, Xu Y, Hurtig HI, Elman L, McMillan CT, Irwin DJ, Porta S, Van Deerlin VM, Lee EB. Annexin A11 aggregation in FTLD-TDP type C and related neurodegenerative disease proteinopathies. Acta Neuropathol. 2024 Jun 19;147(1):104. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Emory University
In the beginning, amyloid was identified by tell-tale morphologic and staining characteristics, and little was known about its constituent proteins. As analytic methods progressed, amyloids became increasingly defined by their principal polymeric proteins, of which more than 40 are now known (Buxbaum et al., 2022). Although amyloid filaments are superficially similar, in recent years, it has become evident that the three-dimensional structure of the filaments formed by a given protein can vary, and that this variation can influence the resulting disease phenotype. Cryo-electron microscopy (cryo-EM) has played a key role in advancing this concept.
While the number of such studies is still small, prior cryo-EM investigations had consistently found that individual amyloid filaments from diseased tissues consisted of a single type of misfolded protein. Various analyses have detected other (usually minor) molecular components of amyloid deposits, but their precise localization and pathogenic importance have mostly been uncertain, and none of them have been shown to be integral constituents of amyloid filaments per se. By demonstrating the coherent integration of two different proteins, annexin-A11 and TDP-43, into individual amyloid filaments from persons with FTLD-TDP Type C, Arseni and colleagues convincingly revise the "one filament, one protein" concept, and raise intriguing questions about amyloidogenesis in general.
The researchers present evidence that annexin-A11 and TDP-43 co-assemble, rather than polymerizing separately into protofilaments that become entwined; does this suggest the presence of heteromeric oligomers, and if so, what are their characteristics? And do heteromeric multimers act as proteopathic seeds, and how might these interact with two separate proteins to induce the formation of heteromeric filaments?
It will be important to determine whether heteromeric filaments occur in other amyloidoses (where they might influence the disease phenotype), or whether the polymers of annexin-A11 and TDP-43 in Type C FTLD-TDP are unique. In this regard, while most auxiliary molecular entities within amyloids probably are not intrinsic to the filaments, revisiting their structural relationship to the filaments with methods such as cryo-EM might shed new light on the ontogeny of the lesions.
The analysis of amyloids by cryo-EM is still in its early stages, yet already it has generated important insights into the surprisingly varied molecular architecture of amyloid filaments. It is gratifying to consider how much we have learned about amyloid and amyloidoses since the advent of the Congo red stain a century ago, but as the elegant work of Arseni and colleagues shows, there is still plenty to learn.
References:
Buxbaum JN, Dispenzieri A, Eisenberg DS, Fändrich M, Merlini G, Saraiva MJ, Sekijima Y, Westermark P. Amyloid nomenclature 2022: update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid. 2022 Dec;29(4):213-219. Epub 2022 Nov 24 PubMed.
University of British Columbia
I think this is a very interesting and exciting discovery for several reasons.
References:
Robinson JL, Suh E, Xu Y, Hurtig HI, Elman L, McMillan CT, Irwin DJ, Porta S, Van Deerlin VM, Lee EB. Annexin A11 aggregation in FTLD-TDP type C and related neurodegenerative disease proteinopathies. Acta Neuropathol. 2024 Jun 19;147(1):104. PubMed.
National Institutes of Health
These parallel discoveries by the Lee and Ryskeldi-Falcon teams are seminal. Together, they conclusively show that two distinct proteins, ANXA11 and TDP-43, co-assemble into organized heteromeric amyloid filaments in FTLD type C and in a subset of other TDP-43 proteinopathies. While previous studies hinted that aggregation-prone proteins could co-localize together in neurodegenerative diseases, most of us assumed that such aggregates were juxtaposed but structurally distinct. That ANXA11 and TDP-43 form interwoven filaments, proven by cryo-EM by the Ryskeldi-Falcon group and quantified pathologically by the Lee group, was entirely unexpected.
These findings further hint at possible upstream origins of the co-filaments, rooted in the normal function of these proteins. Our team previously showed that ANXA11 tethers ribonucleoprotein (RNP) granules to lysosomes for long-distance transport in axons (Liao et al., 2019). At least a fraction of lysosome-associated RNP granules co-traffic with TDP-43, suggesting that ANXA11 and TDP-43 may normally co-assemble in phase condensates during RNA transport. One wonders if the residues bridging ANXA11 and TDP-43 within filamentous assemblies could functionally link these two proteins together within physiological RNPs, but also predispose them to pathological interactions during disease.
There may be a broader role for ANXA11 in neurodegenerative disease than was initially believed. First linked to familial forms of ALS by Chris Shaw’s group, we now know that ANXA11 mutations can cause a range of neurodegenerative diseases. Our team recently found that corticobasal syndrome can be caused by ANXA11 mutations (Snyder et al., 2024), adding to a growing list of clinical syndromes within the ALS/FTD/MSP spectrum associated with mutations in this gene. Interestingly, Eddie Lee saw that approximately 6 percent of non-familial AD/LATE cases had co-pathology of ANXA11-TDP-43 co-aggregates. Given the relatively high prevalence of AD/LATE, this means that ANXA11 pathology is quite common, especially when compared to FTLD type C.
References:
Liao YC, Fernandopulle MS, Wang G, Choi H, Hao L, Drerup CM, Patel R, Qamar S, Nixon-Abell J, Shen Y, Meadows W, Vendruscolo M, Knowles TP, Nelson M, Czekalska MA, Musteikyte G, Gachechiladze MA, Stephens CA, Pasolli HA, Forrest LR, St George-Hyslop P, Lippincott-Schwartz J, Ward ME. RNA Granules Hitchhike on Lysosomes for Long-Distance Transport, Using Annexin A11 as a Molecular Tether. Cell. 2019 Sep 19;179(1):147-164.e20. PubMed.
Snyder A, Ryan VH, Hawrot J, Lawton S, Ramos DM, Qi YA, Johnson KR, Reed X, Johnson NL, Kollasch AW, Duffy MF, VandeVrede L, Cochran JN, Miller BL, Toro C, Bielekova B, Marks DS, Yokoyama JS, Kwan JY, Cookson MR, Ward ME. An ANXA11 P93S variant dysregulates TDP-43 and causes corticobasal syndrome. Alzheimers Dement. 2024 Aug;20(8):5220-5235. Epub 2024 Jun 26 PubMed.
Make a Comment
To make a comment you must login or register.