Gaucher’s Model Recapitulates Phenotype, Supports Drug Candidate
Quick Links
When it comes to studying Gaucher’s disease, a lysosomal storage disorder caused by mutations in the glucocerebrosidase (GBA) gene, good models have been hard to come by. In the June 11 Science Translational Medicine, scientists led by Ellen Sidransky, National Human Genome Research Institute, Bethesda, Maryland, describe a new one that recapitulates the build-up of glycolipids in lysosomes, an important feature of the disease. Using cells from patients with various GBA mutations, the researchers made macrophages—the main cell type affected. Glucocerebrosidase, an enzyme crucial for breakdown of glycolipids, fails in these cells, but its activity was restored by a chaperone molecule that is being examined as a potential therapy. “The model will help us understand more about problem macrophages,” Sidransky told Alzforum. “More importantly, it gives us a way to work on drug development.” The findings have implications beyond Gaucher's, as mutations in the GBA gene are some of the strongest genetic risk factors for Parkinson's disease as well.
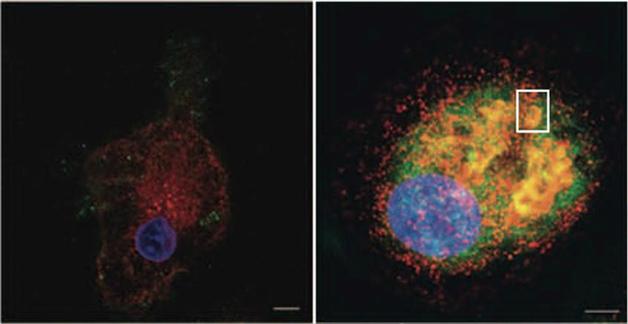
Bring it home. A small molecule refolds and reroutes faulty glucocerebrosidose (green, left), back to lysosomes (red) in treated cells (right). Nucleus appears in blue. See text below. Courtesy of Science Translational Medicine/AAAS.
Macrophages use glucocerebrosidase to break down glycolipids from the membranes of dying white and red blood cells. Patients with Gaucher’s disease have homozygous mutations in GBA that cause glycolipids to build up in the lysosomes. Because these mutations are either lethal in rodents or do not cause a comparable phenotype, no complete mouse models have emerged (see Farfel-Becker et al., 2011). Mouse GBA knockouts do accumulate lipids in lysosomes, but they cannot be used to test treatment strategies that target the mutant protein (see Mistry et al., 2010). Fibroblasts from patients better approximate the disease on the cellular level and are often used for drug screening, but they lack the lysosomal storage problems.
Sidransky’s group previously used patient fibroblasts to find small molecules that cross the blood-brain barrier, act as chaperones to properly fold GBA, and restore its function (see Patnaik et al., 2012). However, without a good model, Sidransky was unable to test whether these drug candidates restore lysosome function.
First author Elma Aflaki and colleagues wanted to see if macrophages from patients with different Gaucher’s genotypes would better model the disease. The researchers cultured macrophages in two ways. First, they took monocytes from the blood of 20 patients with type 1 disease, the kind without neurological pathology. (Types 2 and 3 both have neurological symptoms, with type 2 striking earliest and progressing fastest.) Secondly, they created macrophages from induced pluripotent stem cells (iPSCs) generated from skin cells of five patients with either type 1 or type 2 disease. As controls, they took monocytes from the blood of 30 healthy people and derived iPSCs from one.
In both sets of macrophages, glucocerebrosidase activity measured 3 to 21 percent of control levels. When fed membranes of red blood cells, the macrophages accumulated glucosylceramide and glucosylsphingosine, the same lipids that crowd the lysosomes of patients. While these cells phagocytosed blood cells and bacteria normally, they produced only one-third to one-half of the reactive oxygen species the cells usually use to kill bacteria. What’s more, these cells released fewer of the chemokines that are crucial for migration to sites of inflammation, and they did not wend their way toward chemoattractants as normal macrophages do.
When the researchers treated macrophages with their lead small-molecule chaperone, NCGC00188758, the cells behaved more normally (see image above). Their glucocerebrosidase activity rose 27-fold when presented with erythrocyte membranes. Far fewer glycolipids accumulated, the macrophages began producing normal amounts of reactive oxygen species, and they more readily moved toward chemokines.
“This is a good model for Gaucher disease,” said Pablo Sardi, Genzyme Corporation, Framingham, Massachusetts, who was not involved in the study. Genzyme makes a recombinant glucocerebrosidase that works for patients with type 1 disease, but not for those who have neurological symptoms because the enzyme does not cross into the brain. Sardi cautioned that much work remains to be done with candidate chaperone molecules before any are ready for clinical trials, especially to determine if they will be sufficiently effective to treat Gaucher’s patients.
Sidranksy will continue to use this model to test and improve other candidate chaperones. She conceded that drug development may remain limited without a good animal model, but said she believed that this cellular model would be sufficient to show efficacy while safety can be tested in animals. Sidransky said she next plans to create neurons from Gaucher's disease patient-derived iPSCs and test the chaperone compounds in them, as well.
“This work is important because, so far, fibroblasts have been used for investigating Gaucher’s disease, and they do not accumulate lipids in lysosomes,” said Michela Deleidi, University of Tübingen, Germany. Not only do the macrophages solve that problem, but iPSCs represent a renewable source of cells that can be used for investigating disease pathogenesis and drug screening, she said.
Deleidi recently led a study to compare iPSC-derived neurons from patients with GBA-associated Parkinson’s to such neurons from people with Gaucher’s disease. First author David Schöndorf and colleagues found that glucocerebrosidase activity was weak and α-synuclein and glucosylceramide accumulated in both sets of neurons. Autophagy, a lysosome-based system for degrading unwanted proteins, and intracellular calcium homeostasis were off-balance as well. If the researchers corrected the mutations, all phenotypes reversed, suggesting a causal link. Deleidi said that these neurons will be useful for drug testing, and she plans to try out chaperone therapies. Deleidi hopes that having iPSC lines for the many different known Gaucher’s mutations will allow better personalized medicine for people with the disease.—Gwyneth Dickey Zakaib
References
Paper Citations
- Farfel-Becker T, Vitner EB, Futerman AH. Animal models for Gaucher disease research. Dis Model Mech. 2011 Nov;4(6):746-52. Epub 2011 Oct 4 PubMed.
- Mistry PK, Liu J, Yang M, Nottoli T, McGrath J, Jain D, Zhang K, Keutzer J, Chuang WL, Mehal WZ, Zhao H, Lin A, Mane S, Liu X, Peng YZ, Li JH, Agrawal M, Zhu LL, Blair HC, Robinson LJ, Iqbal J, Sun L, Zaidi M. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc Natl Acad Sci U S A. 2010 Nov 9;107(45):19473-8. PubMed.
- Patnaik S, Zheng W, Choi JH, Motabar O, Southall N, Westbroek W, Lea WA, Velayati A, Goldin E, Sidransky E, Leister W, Marugan JJ. Discovery, structure-activity relationship, and biological evaluation of noninhibitory small molecule chaperones of glucocerebrosidase. J Med Chem. 2012 Jun 28;55(12):5734-48. Epub 2012 Jun 8 PubMed.
External Citations
Further Reading
Papers
- Tiscornia G, Vivas EL, Matalonga L, Berniakovich I, Barragán Monasterio M, Eguizábal C, Gort L, González F, Ortiz Mellet C, García Fernández JM, Ribes A, Veiga A, Izpisua Belmonte JC. Neuronopathic Gaucher's disease: induced pluripotent stem cells for disease modelling and testing chaperone activity of small compounds. Hum Mol Genet. 2013 Feb 15;22(4):633-45. Epub 2012 Oct 31 PubMed.
- Vitner EB, Farfel-Becker T, Eilam R, Biton I, Futerman AH. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher's disease. Brain. 2012 Jun;135(Pt 6):1724-35. Epub 2012 May 7 PubMed.
- Farfel-Becker T, Vitner EB, Kelly SL, Bame JR, Duan J, Shinder V, Merrill AH Jr, Dobrenis K, Futerman AH. Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum Mol Genet. 2014 Feb 15;23(4):843-54. Epub 2013 Sep 24 PubMed.
- Brockmann K, Berg D. The significance of GBA for Parkinson's disease. J Inherit Metab Dis. 2014 Jul;37(4):643-8. Epub 2014 Jun 4 PubMed.
News
Primary Papers
- Aflaki E, Stubblefield BK, Maniwang E, Lopez G, Moaven N, Goldin E, Marugan J, Patnaik S, Dutra A, Southall N, Zheng W, Tayebi N, Sidransky E. Macrophage models of Gaucher disease for evaluating disease pathogenesis and candidate drugs. Sci Transl Med. 2014 Jun 11;6(240):240ra73. PubMed.
- Schöndorf DC, Aureli M, McAllister FE, Hindley CJ, Mayer F, Schmid B, Sardi SP, Valsecchi M, Hoffmann S, Schwarz LK, Hedrich U, Berg D, Shihabuddin LS, Hu J, Pruszak J, Gygi SP, Sonnino S, Gasser T, Deleidi M. iPSC-derived neurons from GBA1-associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun. 2014 Jun 6;5:4028. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.