Inositol Triphosphate Receptor Blamed for Calcium Chaos in Alzheimer’s
Quick Links
Intracellular calcium signaling kicks into overdrive in Alzheimer’s disease neurons, but researchers do not know whether that causes the disease or is the result of it. Researchers from the University of Pennsylvania, Philadelphia, now report that the calcium alterations come first. Led by Kevin Foskett, the scientists scaled back a calcium-release channel in mice expressing human mutant presenilin, reducing calcium release from the endoplasmic reticulum (ER) into the cytosol. The animals generated fewer Aβ plaques in the brain and outsmarted littermates with a full calcium load. The results suggest that this channel, an inositol trisphosphate receptor, has a hand in causing disease driven by presenilin mutations. “This study thoroughly demonstrates the multifaceted role of altered InsP3 receptor function in AD pathology,” said Grace (Beth) Stutzmann, Rosalind Franklin University, North Chicago, Illinois. “It validates how critical and central ER calcium signaling is in the cycle of neurodegeneration.” The results were published May 14 in The Journal of Neuroscience.
Scientists have previously observed too much calcium signaling in fibroblasts and neurons from patients and mice that carry a mutation in presenilin or are overrun by amyloid (see LaFerla 2002; Etcheberrigaray et al., 1998; Jul 2008 news story). While normal calcium release is important for synaptic transmission and plasticity, too much is toxic. Presenilin interacts with the type 1 inositol trisphosphate receptor (InsP3R1) in the ER. Foskett and colleagues previously found that mutant presenilin boosted the activity of the InsP3R1 channel (see Cheung et al., 2008), and then wondered if this revved-up calcium signaling drove Alzheimer’s pathology.
To find out, first author Dustin Shilling and colleagues genetically halved InsP3R1 in AD mouse models by breeding them with the heterozygous Opisthotonos (Opt) mouse (Street et al., 1997). The homozygote version of this animal is severely epileptic as it lacks two functional copies of InsP3R1, but heterozygotes have a milder phenotype. Crossing Opt mice with two models that express mutated presenilin—the M164V knock-in and 3xTg mice—brought calcium signaling back down to wild-type levels in the double transgenics.
The researchers first looked in the M146V/Opt cross. Because these mice develop no plaques or tangles, the researchers could observe the effects of reduced calcium signaling without interference from amyloid. In primary cultured neurons and hippocampal slices from these mice, long-term potentiation and transcriptional pathways activated by calcium release were normal compared to the M146 knock-ins. These results suggested that correcting the calcium imbalance restored normal electrophysiology and gene expression.
Would less calcium signaling also lower AD pathology? To answer that question, the group examined the 3xTg/Opt mice. Halving InsP3R1 reduced Aβ and phosphorylated tau in the hippocampus and cortex.
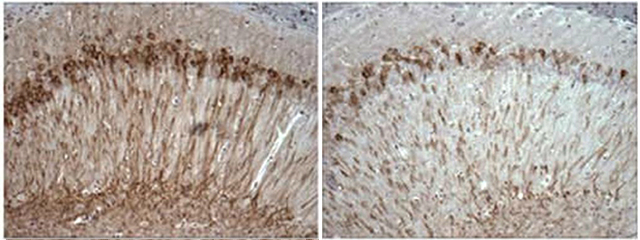
Reduced calcium signaling (right) curbs accumulation of phosphorylated tau (brown) in the hippocampus of the 3xTg mouse. [Image courtesy of Foskett et al., The Journal of Neuroscience, 2014. Used with permission.]
The halving of InsP3R1 also rescued the impaired LTP typically seen in hippocampal slices from 3xTG animals. 3xTg/Opt mice remembered objects as well as wild-type mice and froze as often as controls in a contextual fear-conditioning task. These data suggest that too much calcium signaling leads to the pathology and behavioral symptoms of AD that arise from presenilin mutations. “The results were profound,” said Foskett. “This is likely a factor that contributes to the pathogenesis of familial Alzheimer’s in patients with presenilin mutations,” he said.
Foskett said his group will next create mice in which the InsP3R1 allele can be turned down at various ages, to see how early calcium signaling must be modified to stave off or treat disease. He also plans to figure out how to target this receptor with drugs. InsP3R1 is present in most cells of the body, but the lack of adverse effects in heterozygote InsP3R1 mice hints that safely modifying it may be possible, he said.
“The study suggests that inhibition of InsP3 receptors may have some benefit in AD,” said Ilya Bezprozvanny, University of Texas Southwestern Medical Center, Dallas. He cautioned that because mice homozygous for a defunct InsP3R1 allele are profoundly epileptic, pharmaceutical companies may shy away from pursuing the receptor as a drug target. Bezprozvanny and colleagues recently found that knocking down the ryanodine receptor type 3—the other major receptor to release calcium from the neuronal ER—in older APPPS1 mice led to fewer AD symptoms, reinforcing ER calcium channels as important in AD (see Liu et al., 2014). However, like InsP3R1, ryanodine receptors occur throughout the body, so modifying them could lead to side effects. To specifically target neurons, Bezprozvanny’s group has proposed modulating calcium influx through channels in dendritic spines (see Apr 2014 news story).
For her part, Stutzmann is testing drugs that tweak malfunctioning ryanodine receptors in AD mouse models. Mark Mattson, National Institute on Aging, Baltimore, cited efforts to stimulate inhibitory neurons (see Aug 2012 news story). Doing so prevents toxic calcium release and could be a way to restrain calcium signaling indirectly. To find out whether the current study extrapolates to sporadic forms of AD, researchers could cross Opt mice with those that have Aβ pathology but no PS1 mutation, he proposed.—Gwyneth Dickey Zakaib
References
News Citations
- More Calcium News: Plaques Cause Dendrite Damage via Ion Overload
- Calcium Sensor STIM2 Maintains Synapses, Ebbs in Alzheimer’s
- Anticonvulsants Reverse AD-like Symptoms in Transgenic Mice
Research Models Citations
Paper Citations
- Laferla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002 Nov;3(11):862-72. PubMed.
- Etcheberrigaray R, Hirashima N, Nee L, Prince J, Govoni S, Racchi M, Tanzi RE, Alkon DL. Calcium responses in fibroblasts from asymptomatic members of Alzheimer's disease families. Neurobiol Dis. 1998 Jul;5(1):37-45. PubMed.
- Cheung KH, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008 Jun 26;58(6):871-83. PubMed.
- Street VA, Bosma MM, Demas VP, Regan MR, Lin DD, Robinson LC, Agnew WS, Tempel BL. The type 1 inositol 1,4,5-trisphosphate receptor gene is altered in the opisthotonos mouse. J Neurosci. 1997 Jan 15;17(2):635-45. PubMed.
- Liu J, Supnet C, Sun S, Zhang H, Good L, Popugaeva E, Bezprozvanny I. The role of ryanodine receptor type 3 in a mouse model of Alzheimer disease. Channels (Austin). 2014 Jan 29;8(3) PubMed. Expression of Concern.
Further Reading
Papers
- Jensen LE, Bultynck G, Luyten T, Amijee H, Bootman MD, Roderick HL. Alzheimer's disease-associated peptide Aβ42 mobilizes ER Ca(2+) via InsP3R-dependent and -independent mechanisms. Front Mol Neurosci. 2013;6:36. PubMed.
- Oulès B, Del Prete D, Greco B, Zhang X, Lauritzen I, Sevalle J, Moreno S, Paterlini-Bréchot P, Trebak M, Checler F, Benfenati F, Chami M. Ryanodine receptor blockade reduces amyloid-β load and memory impairments in Tg2576 mouse model of Alzheimer disease. J Neurosci. 2012 Aug 22;32(34):11820-34. PubMed.
- Chakroborty S, Briggs C, Miller MB, Goussakov I, Schneider C, Kim J, Wicks J, Richardson JC, Conklin V, Cameransi BG, Stutzmann GE. Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer's disease. PLoS One. 2012;7(12):e52056. PubMed.
- Stutzmann GE, Mattson MP. Endoplasmic reticulum Ca(2+) handling in excitable cells in health and disease. Pharmacol Rev. 2011 Sep;63(3):700-27. PubMed.
- Mattson MP. ER calcium and Alzheimer's disease: in a state of flux. Sci Signal. 2010;3(114):pe10. PubMed.
Primary Papers
- Shilling D, Müller M, Takano H, Mak DO, Abel T, Coulter DA, Foskett JK. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer's disease pathogenesis. J Neurosci. 2014 May 14;34(20):6910-23. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.