Introducing: iPSC Collection from Tauopathy Patients
Quick Links
A multi-institutional group, including members of the Tau Consortium, unveiled a stem cell tool kit for scientists studying primary tauopathies. In the November 12 issue of Stem Cell Reports, researchers co-led by Celeste Karch of Washington University, St. Louis, and Alison Goate and Sally Temple of Icahn School of Medicine in New York, describe a collection of fibroblasts, induced pluripotent stem cells, and neural precursor cells. The cells come from 140 skin samples, some given by donors with richly documented clinical histories who carry pathogenic MAPT mutations or risk variants. Others come from noncarrier family members, patients with a sporadic tauopathy, and cognitively normal controls. The set includes induced pluripotent stem cell lines from 31 donors and 21 CRISPR-engineered isogenic lines. The cells are available to other researchers for study.
- Multi-institutional group announces primary tauopathy stem cell resource.
- It includes fibroblasts, iPSCs, and NPCs from MAPT mutation carriers and individuals with sporadic disease, with deep clinical histories.
- It also includes 21 CRISPR-edited isogenic lines.
“These types of high-quality repositories are becoming increasingly important for the scientific community,” Clive Svendsen of the Cedars-Sinai Medical Center in Los Angeles wrote to Alzforum.
“This is the way the field is going,” agreed Lawrence Golbe of CurePSP, New York. Golbe’s organization funds research into progressive nuclear palsy (PSP) and related disorders, and collaborates with the Tau Consortium on other projects. Enthusiastic about the resource’s potential, Golbe hopes CurePSP grantees will get an automatic pass to use the cells.
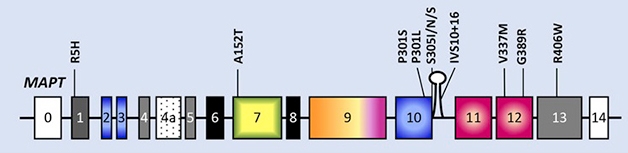
Choice Mutations. Cells in the new iPSC collection carry some of the most common MAPT mutations, covering a wide range of clinical and neuropathological phenotypes of frontotemporal lobe dementia (FTLD)-Tau. [Courtesy of Karch et al., 2019.]
Tauopathies have proven difficult to study in animal models, in part because unlike other neuropathologies, they seem to afflict only humans (Heuer et al., 2012). Moreover, while adult human brains express approximately equal amounts of the tau spliced isoforms 3R and 4R, rodents produce almost exclusively 4R (Trabzuni et al., 2012). This is problematic. For example, leading proposals to explain how tau mutations cause disease point to abnormalities in splicing and microtubule binding, which differ between isoforms. “The models we had been focusing on were not capturing the complexity of MAPT in human cells,” said first author Karch. As a result, human induced pluripotent stem cells (iPSCs) have been gaining popularity in the field. The NINDS Human Cell and Data Repository is helping meet the demand by offering iPSC lines derived from 10 patients harboring MAPT mutations.
However, Karch and her collaborators think the field could benefit from a larger and more diverse collection of human cells, including isogenic iPSC lines. To accomplish this, they collected skin samples from 140 people carrying MAPT pathogenic mutations or risk variants, non-mutation carriers, and patients with sporadic PSP or corticobasal syndrome (CBS), most with comprehensive clinical histories. Although a few cells came from the NINDS repository, most came from patients participating in longitudinal studies at the Memory and Aging Center at the University of California, San Francisco, and the Knight Alzheimer Disease Research Center at WashU. The clinical records of most of these patients include detailed neurological and neuropathological workups, as well as fluid biomarkers and neuroimaging data collected from MRI, Aβ-PET, and tau-PET studies.
To capture a broad range of phenotypes associated with some of the most common MAPT mutations, the authors created 36 fibroblast lines and 29 iPSC lines from individuals carrying the P301L, S305I, IVS10+16, V337M, G389R, and R406W mutations, as well as from carriers of the A152T variant, which increases the risk for both PSP and CBS (image above). The latter could be particularly useful for dissecting the mechanisms that underlie the phenotypic differences between the two diseases. The researchers also obtained iPSC lines from two noncarrier family members, and two people who suffered from autopsy-confirmed sporadic PSP. In addition, they stored fibroblast lines from 12 patients with sporadic PSP, five with CBS, 10 with a mixed PSP/CBS presentation, and 69 cognitively normal controls.
Biopsies are available for 27 of the 31 patients whose cells were used to generate iPSCs, and autopsy data for seven, including the two cases of sporadic PSP.
Importantly, the researchers edited 21 iPSC lines using CRISPR/Cas 9. They corrected cells with these mutations: MAPT IVS10+16, P301L, S305I, R406W, and V337M. Conversely, they inserted into control iPSCs these mutations: R5H, P301L, G389R, S305I, or S305S.
The authors also created a stem cell line carrying MAPT P301S, a mutation commonly overexpressed in tauopathy mouse models but not present in the available donors, by editing the P301L line. “Isogenic lines are so powerful, particularly in these diseases which are so variable in their onset and progression, even within the same family,” said Karch. Günter Höglinger and Tabea Strauss at the German Center for Neurodegenerative Disease (DZNE) in Munich agreed. “Having a pool of cell lines with different disease-linked mutations and risk variants from several individuals and their isogenic control cells is an excellent resource for the research community to enlighten disease mechanisms,” they wrote (full comment below).
Several of the reported lines have already starred in recent studies of tauopathy mechanisms and candidate therapies (e.g., Sep 2019 conference news; Nakamura et al., 2019; Hernandez et al., 2019; Silva et al., 2019).
Karch and colleagues have partially differentiated some of the iPSCs and stored them as neural progenitor cells (NPCs), so that researchers can relatively easily thaw, expand, and differentiate them into neurons. These NPCs have proved useful for large-scale functional-genomics studies, proteomics, and genetic modifier screens (e.g., Cheng et al., 2017; Boselli et al., 2017; Tian et al., 2019).
In addition, the authors inserted a neurogenin-2 transgene into two healthy controls and two MAPT mutant stem cells, P301L and R406W. Neurogenin-2 enables low-cost, large-scale differentiation of stem cells into homogenous excitatory neurons. These transgenic cells are particularly useful for high-throughput drug screens (Wang et al., 2017; Sohn et al., 2019).
Researchers can request all the reported cells online at http://neuralsci.org/tau. They must provide a summary of experimental plans, an institutional material transfer agreement, and a nominal fee to cover maintenance and distribution costs. Karch said the process resembles that of the Coriell Institute and the NINDS repository. “Our goal is to share with as few hurdles as possible,” she said.
While the authors are still reprogramming fibroblasts they have already collected, they also plan to add more causative mutations, generate more isogenic lines, and obtain more cells from members of the same families to help shed light on phenotypic variability. In addition, Karch said, she hopes repository users will resubmit lines with new modifications they generate.
Jeffrey Rothstein, Johns Hopkins University, Baltimore, welcomed the new resource. “I think it is great they have assembled this collection,” he said. Rothstein founded and co-directs the Answer ALS research project, which has amassed 600 iPSC lines from controls and patients with amyotrophic lateral sclerosis (ALS).
Rothstein suggested the tauopathy collection may want to prioritize adding cells from donors with the most common form of disease, that is, sporadic. His group aims to generate 1,000 iPSC lines, with a large fraction representing sporadic disease—also the most common form of ALS—to identify the most prevalent disease subtypes. One strategy that has helped his group build their collection, he said, is using peripheral blood mononuclear cells instead of fibroblasts to create iPSCs. More donors are willing to donate blood than have a piece of skin punched out. In addition, iPSCs derived from blood cells are genetically more stable, he noted.
Rothstein emphasized the importance of assembling a large collection of healthy controls. Although isogenic controls are of great value, he cautioned they can be subject to artifacts. One problem is that the cell population can change due to selective pressures during CRISPR editing (Budde et al., 2017). To address this, Karch and colleagues are collecting not only modified iPSC clones, but also control clones that have gone through the editing pipeline but remain unmodified.
Stem-cell users studying tauopathies face another challenge: iPSC-derived neurons express primarily the fetal isoform of tau, 3R0N. However, citing a study that shows three-dimensional neuronal cultures switch to the adult profile relatively quickly (Miguel et al., 2019), Höglinger and Strauss wrote, “[It] allows us to be optimistic that current challenges of this model system can be overcome in the future.”—Marina Chicurel
References
Mutations Citations
- MAPT P301L
- MAPT S305I
- MAPT IVS10+16 C>T
- MAPT V337M
- MAPT R406W
- MAPT A152T
- MAPT R5H
- MAPT S305S
- MAPT P301S
Mutation Position Table Citations
News Citations
Paper Citations
- Heuer E, Rosen RF, Cintron A, Walker LC. Nonhuman primate models of Alzheimer-like cerebral proteopathy. Curr Pharm Des. 2012;18(8):1159-69. PubMed.
- Trabzuni D, Wray S, Vandrovcova J, Ramasamy A, Walker R, Smith C, Luk C, Gibbs JR, Dillman A, Hernandez DG, Arepalli S, Singleton AB, Cookson MR, Pittman AM, de Silva R, Weale ME, Hardy J, Ryten M. MAPT expression and splicing is differentially regulated by brain region: relation to genotype and implication for tauopathies. Hum Mol Genet. 2012 Sep 15;21(18):4094-103. Epub 2012 Jun 20 PubMed.
- Nakamura M, Shiozawa S, Tsuboi D, Amano M, Watanabe H, Maeda S, Kimura T, Yoshimatsu S, Kisa F, Karch CM, Miyasaka T, Takashima A, Sahara N, Hisanaga SI, Ikeuchi T, Kaibuchi K, Okano H. Pathological Progression Induced by the Frontotemporal Dementia-Associated R406W Tau Mutation in Patient-Derived iPSCs. Stem Cell Reports. 2019 Oct 8;13(4):684-699. Epub 2019 Sep 19 PubMed.
- Hernandez I, Luna G, Rauch JN, Reis SA, Giroux M, Karch CM, Boctor D, Sibih YE, Storm NJ, Diaz A, Kaushik S, Zekanowski C, Kang AA, Hinman CR, Cerovac V, Guzman E, Zhou H, Haggarty SJ, Goate AM, Fisher SK, Cuervo AM, Kosik KS. A farnesyltransferase inhibitor activates lysosomes and reduces tau pathology in mice with tauopathy. Sci Transl Med. 2019 Mar 27;11(485) PubMed.
- Silva MC, Ferguson FM, Cai Q, Donovan KA, Nandi G, Patnaik D, Zhang T, Huang HT, Lucente DE, Dickerson BC, Mitchison TJ, Fischer ES, Gray NS, Haggarty SJ. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife. 2019 Mar 25;8 PubMed.
- Boselli M, Lee BH, Robert J, Prado MA, Min SW, Cheng C, Silva MC, Seong C, Elsasser S, Hatle KM, Gahman TC, Gygi SP, Haggarty SJ, Gan L, King RW, Finley D. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J Biol Chem. 2017 Nov 24;292(47):19209-19225. Epub 2017 Sep 26 PubMed.
- Tian R, Gachechiladze MA, Ludwig CH, Laurie MT, Hong JY, Nathaniel D, Prabhu AV, Fernandopulle MS, Patel R, Abshari M, Ward ME, Kampmann M. CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron. 2019 Oct 23;104(2):239-255.e12. Epub 2019 Aug 15 PubMed.
- Wang C, Ward ME, Chen R, Liu K, Tracy TE, Chen X, Xie M, Sohn PD, Ludwig C, Meyer-Franke A, Karch CM, Ding S, Gan L. Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Reports. 2017 Oct 10;9(4):1221-1233. Epub 2017 Sep 28 PubMed.
- Sohn PD, Huang CT, Yan R, Fan L, Tracy TE, Camargo CM, Montgomery KM, Arhar T, Mok SA, Freilich R, Baik J, He M, Gong S, Roberson ED, Karch CM, Gestwicki JE, Xu K, Kosik KS, Gan L. Pathogenic Tau Impairs Axon Initial Segment Plasticity and Excitability Homeostasis. Neuron. 2019 Nov 6;104(3):458-470.e5. Epub 2019 Sep 18 PubMed.
- Budde JP, Martinez R, Hsu S, Wen N, Chen JA, Coppola G, Goate AM, Cruchaga C, Karch CM. Precision genome-editing with CRISPR/Cas9 in human induced pluripotent stem cells. bioRxiv September 12, 2017. BioRxiv.
- Miguel L, Rovelet-Lecrux A, Feyeux M, Frebourg T, Nassoy P, Campion D, Lecourtois M. Detection of all adult Tau isoforms in a 3D culture model of iPSC-derived neurons. Stem Cell Res. 2019 Oct;40:101541. Epub 2019 Aug 23 PubMed.
Other Citations
External Citations
Further Reading
Papers
- Hallmann AL, Araúzo-Bravo MJ, Mavrommatis L, Ehrlich M, Röpke A, Brockhaus J, Missler M, Sterneckert J, Schöler HR, Kuhlmann T, Zaehres H, Hargus G. Astrocyte pathology in a human neural stem cell model of frontotemporal dementia caused by mutant TAU protein. Sci Rep. 2017 Mar 3;7:42991. PubMed.
- Imamura K, Sahara N, Kanaan NM, Tsukita K, Kondo T, Kutoku Y, Ohsawa Y, Sunada Y, Kawakami K, Hotta A, Yawata S, Watanabe D, Hasegawa M, Trojanowski JQ, Lee VM, Suhara T, Higuchi M, Inoue H. Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci Rep. 2016 Oct 10;6:34904. PubMed.
- Seo J, Kritskiy O, Watson LA, Barker SJ, Dey D, Raja WK, Lin YT, Ko T, Cho S, Penney J, Silva MC, Sheridan SD, Lucente D, Gusella JF, Dickerson BC, Haggarty SJ, Tsai LH. Inhibition of p25/Cdk5 Attenuates Tauopathy in Mouse and iPSC Models of Frontotemporal Dementia. J Neurosci. 2017 Oct 11;37(41):9917-9924. Epub 2017 Sep 14 PubMed.
Primary Papers
- Karch CM, Kao AW, Karydas A, Onanuga K, Martinez R, Argouarch A, Wang C, Huang C, Sohn PD, Bowles KR, Spina S, Silva MC, Marsh JA, Hsu S, Pugh DA, Ghoshal N, Norton J, Huang Y, Lee SE, Seeley WW, Theofilas P, Grinberg LT, Moreno F, McIlroy K, Boeve BF, Cairns NJ, Crary JF, Haggarty SJ, Ichida JK, Kosik KS, Miller BL, Gan L, Goate AM, Temple S, Tau Consortium Stem Cell Group. A Comprehensive Resource for Induced Pluripotent Stem Cells from Patients with Primary Tauopathies. Stem Cell Reports. 2019 Nov 12;13(5):939-955. Epub 2019 Oct 17 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
DTx Pharma
In this paper, we are witness to a momentous birth announcement, presenting the work of a talented, multidisciplinary group of scientists focused on tauopathies. Over at least the past two decades, the tau protein and Tau gene have been studied first as phenotypic markers and increasingly as both genetic and prion-like drivers of neurodegenerative diseases, including some types of FTLD, PSP, CBD, and AD. Terrific insights have come from forced overexpression models employing mutant Tau (PS19 mice, for instance) in vivo and in vitro, where toxicity of abnormal tau protein species (mutant or misfolded WT) have been characterized. Even so, the relevance of these model systems to critical drivers of pathogenesis in patients, and their utility as the generator of phenotypes to be modified in drug screens, remains uncertain. We’re just beginning to read out the first therapeutic candidates that were screened using these legacy models as part of their preclinical proof-of-concept packages.
This paper describes the initial set of fibroblasts, iPS cells, and neural progenitor cells derived from FTLD/PSP/CBD patients now available to both basic scientists and drug developers who wish to target Tau-mediated disorders. The resource includes a number of distinct, known pathogenic Tau mutations derived from well-characterized patients/families. Importantly, the resource includes several isogenic lines that differ only in regard to the presence or absence of a particular, pathogenic tau mutation—effectively allowing researchers to control for differences in genetic background or modifier genes that might otherwise cloud interpretation of experimental results.
Two other critical points are worth emphasizing. First, some lines were derived from patients without Tau mutations. Because most cases of PSP and CBD are “sporadic” and do not harbor Tau mutations, study of these lines may provide additional information on modifier genes that affect tau production or elimination. Second, the consortium that developed this resource continue to generate additional lines for study of Tau-mediated disorders.
Patients, families, researchers, and companies are enthusiastic about accessing these new lines and appreciate the open access provided by the researchers and funding organizations that have supported this initiative.
Deutsches Zentrum für Neurodegenerative Erkrankungen e.V. (DZNE)
Hannover Medical School
The importance of iPSC resource for primary tauopathies
Tauopathies are a group of neurodegenerative diseases representing pathological inclusions composed of aggregates and modified forms of microtubule-associated protein Tau (MAPT). Tau inclusions can co-exist with other pathological protein aggregates such as Aβ in Alzheimer’s disease. However, Tau aggregates can also be the primary factor driving neurodegeneration in diseases classified as primary tauopathies. The latter can be sporadic or associated with mutations in the MAPT gene predisposing for Tau pathology. Tau pathology is explained via two main mechanisms. First, aggregates of Tau protein may disrupt the cellular process to cause neuronal degeneration (gain of toxic function theory). Secondly, Tau post-translational modifications (e.g. phosphorylation) may interfere with the physiological function of Tau as a microtubule-binding protein, leading to microtubule destabilization and consequently impairment of axonal transport (loss-of-function theory).
Despite all knowledge obtained on Tau-dependent neurodegeneration in the past decades, there is still no approved therapy for tauopathies. Many aspects of Tau pathology are still unknown, such as the trigger factors for pathology, the differential sensitivity of cells toward tau pathology, and the cellular and molecular mechanisms of Tau propagation. Studying the biological consequences of Tau mutations in models of primary tauopathies provides a unique opportunity to decipher disease mechanisms and to develop Tau-targeting therapies.
A big challenge remains to create a model system that is as close as possible to the human disease. Patient-derived cell models are still in the process of optimization, but a major step forward has been achieved by Karch and colleagues by establishing a comprehensive resource for induced pluripotent stem cell (iPSC) lines from patients with primary tauopathies.
This resource of iPSC lines is therefore an enormously valuable asset in the research on Tau-related neurodegenerative diseases. Having a pool of cell lines with different disease-linked mutations and risk variants from several individuals and their isogenic control cells is an excellent resource for the research community to enlighten disease mechanisms. Different mutations in the MAPT gene have different effects on the Tau protein. While some mutations have been associated with a changed 3R/4R Tau ratio (Liu and Gong, 2008), others are known to alter the protein sequence (Fischer et al., 2007). Therefore, the variety of available cell lines and their mutations will help to model the different aspects of relevant disease mechanisms. In addition, having detailed clinical data for most of the individuals from whom the cell lines were generated is a major advance.
The current limitation of iPSC models is their relative immaturity in terms of neuronal differentiation. Without an extensive culturing period, only the fetal Tau isoform is expressed in iPSC-derived neurons (Sposito et al., 2015). Since quite a number of mutations in the MAPT gene effect the 4R Tau isoform splicing, this issue is of crucial relevance. Improvements in differentiation protocols are needed to speed up the maturation process and induce a switch to a Tau isoform profile more closely resembling the adult brain. Recent findings (Miguel et al., 2019) show that three-dimensional culturing of neurons can reduce the differentiation time until all six Tau isoforms are expressed in iPSC and allow us to be optimistic that current challenges of this model system can be overcome in the future.
References:
Fischer D, Mukrasch MD, von Bergen M, Klos-Witkowska A, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M. Structural and microtubule binding properties of tau mutants of frontotemporal dementias. Biochemistry. 2007 Mar 13;46(10):2574-82. PubMed.
Liu F, Gong CX. Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener. 2008 Jul 10;3:8. PubMed.
Miguel L, Rovelet-Lecrux A, Feyeux M, Frebourg T, Nassoy P, Campion D, Lecourtois M. Detection of all adult Tau isoforms in a 3D culture model of iPSC-derived neurons. Stem Cell Res. 2019 Oct;40:101541. Epub 2019 Aug 23 PubMed.
Sposito T, Preza E, Mahoney CJ, Setó-Salvia N, Ryan NS, Morris HR, Arber C, Devine MJ, Houlden H, Warner TT, Bushell TJ, Zagnoni M, Kunath T, Livesey FJ, Fox NC, Rossor MN, Hardy J, Wray S. Developmental regulation of tau splicing is disrupted in stem cell-derived neurons from frontotemporal dementia patients with the 10 + 16 splice-site mutation in MAPT. Hum Mol Genet. 2015 Sep 15;24(18):5260-9. Epub 2015 Jul 1 PubMed.
Make a Comment
To make a comment you must login or register.