A Kinder Pathogen? Viral Protein Preserves Neurons in Parkinson’s Model
Quick Links
Could infectious agents hold the key to treating neurodegenerative disease? The idea may not be as crazy as it sounds. Some neuropathogens have evolved ways to turn off cell death mechanisms to keep their hosts alive. The same approach might rescue sick neurons in diseased brains, according to a paper in the 21 October Nature Communications. Researchers led by Daniel Gonzalez-Dunia at INSERM in Toulouse, France, reported that the anonymous-sounding protein X from the Borna disease virus protected axons and neurons from degeneration in a mouse model of Parkinson’s disease. Protein X, as well as a peptide derived from it, bolstered mitochondrial function in the stressed cells. The strategy might work for other disorders characterized by mitochondrial dysfunction, such as amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease, suggest the researchers. “This peptide serves as a proof of concept that opens the way for designing new neuroprotective drugs,” Gonzalez-Dunia told Alzforum.
“I found the paper very intriguing,” said Brian Balin at the Philadelphia College of Osteopathic Medicine, Pennsylvania. “It offers a lead into how you could combat neurodegeneration.” Balin, who was not involved in the research, has studied links between infectious agents and dementia.
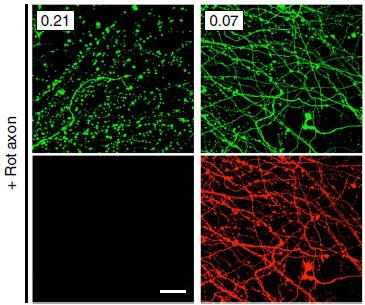
Axon Protector.
After application of rotenone, a mitochondrial toxin, axons (green) degenerated (left panels), but not when the cells were infected (right panels) with BDV (red). [Image courtesy of Nature Communications and Szelechowski et al., 2014.]
Borna disease virus (BDV) is an RNA virus that occurs throughout the world and mostly infects animals, particularly horses and sheep. In these species, the disease hampers movement, behavior, and learning, and often leads to death. People can harbor the virus as well, but researchers disagree on whether the infection leads to any visible symptoms. Some studies have linked BDV to neuropsychiatric disorders, but others refute this connection (see, e.g., Miranda et al., 2006; Hornig et al., 2012). The virus integrates into neuronal DNA, where it persists for years. In cultured glial cells, the virus’ protein X clings to mitochondria and prevents apoptosis (see Poenisch et al., 2009; Li et al., 2013).
Gonzalez-Dunia and colleagues wondered if BDV could protect neurons from nonviral stressors as well. First author Marion Szelechowski infected cultured primary neurons with BDV, then applied the toxin rotenone to their axons. Rotenone disrupts the mitochondrial electron transport chain, and in control cultures causes axons to gradually die back to the cell body. However, BDV-infected axons remained intact (see image above). Since previous work had flagged protein X as anti-apoptotic, the authors next applied a recombinant version to fresh cultures, and found that the protein alone protected axons from rotenone as well as did BDV infection.
Because rotenone causes a Parkinson-like disorder in rodents, the researchers wanted to see if protein X also protected neurons in a mouse model of PD. They injected a lentivirus expressing the protein into the substantia nigra of mice two weeks prior to treating them with the neurotoxin MPTP, which, like rotenone, targets mitochondria and kills dopaminergic neurons. As a control, they used a mutated version of the protein that lacked a mitochondrial localization sequence. Three weeks after MPTP administration, control animals had lost about a third of their dopamine neurons, while those receiving protein X maintained them.
While the findings suggested protein X might have therapeutic potential, the researchers felt the viral delivery method was cumbersome. The authors examined various domains of the protein to see if they could find a fragment that retained activity and could be more easily administered. They found that the C-terminal third, with a mitochondrial localization tag added, provided as much protection as full protein X. This small peptide, which they called PX3, could be administered intranasally, achieving high levels in the striatum and distributing throughout the brain and spinal cord, as well as in some peripheral organs. Mice who sniffed up the spray a day before MPTP injection retained 40 percent more dopamine neurons than control animals.
How do protein X and PX3 work? Mitochondria in treated cells grew up to four times longer than those in control neurons. This probably results from fusion of mitochondria, and represents a response to cellular stress, Gonzalez-Dunia told Alzforum. The massive mitochondria produce more energy and may dilute apoptotic signals, allowing cells to survive, he suggested.
Normally, dynamin-related protein 1 (Drp1) promotes fission of elongated mitochondria. In PX3-treated cells, however, active, phosphorylated Drp1 dropped by half. PX3 probably acts indirectly on Drp1, the authors concluded, since they found no evidence that the two molecules bind. Interestingly, in Alzheimer’s disease, and after rotenone treatment, Drp1 becomes overactive. This causes mitochondria to fragment, precipitating cell death (see Apr 2009 news story). Too little Drp1 also creates problems; a recent study reported that deletion of the protein in mouse dopamine neurons led to loss of mitochondria, axons, and neurons (Berthet et al., 2014).
Gonzalez-Dunia's data suggest that PX3 protects neurons in a preventative model. Could the peptide also treat ongoing disease? He is investigating this in animals that model chronic, progressive Parkinson’s disease. Because mitochondrial dysfunction characterizes many neurodegenerative disorders, he is also testing PX3 in models of Alzheimer’s and ALS (see Mar 2013 news story; Oct 2014 news story; Oct 2014 news story).
While the consequences of chronic administration of PX3 are unclear, treated mice show no ill effects. Before MPTP administration, neurons maintained normal cellular respiration and had few elongated mitochondria, Gonzalez-Dunia said. “Under normal conditions, cells are not affected by the peptide, but in response to stress, the mitochondria are better armed to respond,” he told Alzforum. He plans to further study the peptide’s pharmacodynamics and potential toxicity. He noted that PX3 itself might not become a therapy; instead, it might point the way toward better peptides or drugs that target the same mechanism.
The protective abilities of BDV and protein X may come as a surprise, since other work suggests pathogens increase risk for neurodegenerative disease (see, e.g., Oct 2004 conference story; Aug 2009 news story). Researchers led by Fredrik Elgh at Umeå University, Sweden, recently found that people infected with the Herpes simplex virus for seven years or more, and those with active Herpes simplex infections, run double the risk of developing AD (see also Feb 2011 webinar). The results appear in two papers in the October Alzheimer’s and Dementia. These data support earlier research by Ruth Itzhaki at the University of Manchester, U.K., who has long championed the idea of a link between HSV infection and AD (see Jun 2004 webinar; Wozniak et al., 2009).
Likewise, Balin has linked infection with the respiratory bacterium Chlamydia pneumoniae to Alzheimer’s onset (see Little et al., 2004; Gérard et al., 2006). Intriguingly, Chlamydia infections also dial down neuronal apoptosis and lead to misshapen, enlarged mitochondria, Balin told Alzforum (see Appelt et al., 2008). He plans to investigate whether Chlamydia makes a protein analogous to BDV’s protein X. “Maybe there is a global [anti-apoptotic] mechanism common to persistent infections in the nervous system,” he speculated.—Madolyn Bowman Rogers
References
News Citations
- NO Kidding? Mitochondria Fission Protein Linked to Neurodegeneration
- Protein Destroying Muscle, Bone, Nerves Parks on Mitochondria
- Evidence Mounts That Mitochondrial Gene Is Bona Fide ALS, FTD Risk Factor
- ALS, Parkinson’s Proteins Co-Mingle in Mitochondria Destruction Pathway
- San Diego: HIV and AD—Save the Body, Lose the Mind?
- Bugs on the Brain—Can Flu Cause Parkinsonism, Neurodegeneration?
Webinar Citations
Paper Citations
- Miranda HC, Nunes SO, Calvo ES, Suzart S, Itano EN, Watanabe MA. Detection of Borna disease virus p24 RNA in peripheral blood cells from Brazilian mood and psychotic disorder patients. J Affect Disord. 2006 Jan;90(1):43-7. Epub 2005 Dec 1 PubMed.
- Hornig M, Briese T, Licinio J, Khabbaz RF, Altshuler LL, Potkin SG, Schwemmle M, Siemetzki U, Mintz J, Honkavuori K, Kraemer HC, Egan MF, Whybrow PC, Bunney WE, Lipkin WI. Absence of evidence for bornavirus infection in schizophrenia, bipolar disorder and major depressive disorder. Mol Psychiatry. 2012 May;17(5):486-93. Epub 2012 Jan 31 PubMed.
- Poenisch M, Burger N, Staeheli P, Bauer G, Schneider U. Protein X of Borna disease virus inhibits apoptosis and promotes viral persistence in the central nervous systems of newborn-infected rats. J Virol. 2009 May;83(9):4297-307. Epub 2009 Feb 11 PubMed.
- Li Y, Song W, Wu J, Zhang Q, He J, Li A, Qian J, Zhai A, Hu Y, Kao W, Wei L, Zhang F, Xu D. MAVS-mediated host cell defense is inhibited by Borna disease virus. Int J Biochem Cell Biol. 2013 Aug;45(8):1546-55. Epub 2013 May 20 PubMed.
- Berthet A, Margolis EB, Zhang J, Hsieh I, Zhang J, Hnasko TS, Ahmad J, Edwards RH, Sesaki H, Huang EJ, Nakamura K. Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J Neurosci. 2014 Oct 22;34(43):14304-17. PubMed.
- Wozniak MA, Mee AP, Itzhaki RF. Herpes simplex virus type 1 DNA is located within Alzheimer's disease amyloid plaques. J Pathol. 2009 Jan;217(1):131-8. PubMed.
- Little CS, Hammond CJ, MacIntyre A, Balin BJ, Appelt DM. Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol Aging. 2004 Apr;25(4):419-29. PubMed.
- Gérard HC, Dreses-Werringloer U, Wildt KS, Deka S, Oszust C, Balin BJ, Frey WH, Bordayo EZ, Whittum-Hudson JA, Hudson AP. Chlamydophila (Chlamydia) pneumoniae in the Alzheimer's brain. FEMS Immunol Med Microbiol. 2006 Dec;48(3):355-66. PubMed.
- Appelt DM, Roupas MR, Way DS, Bell MG, Albert EV, Hammond CJ, Balin BJ. Inhibition of apoptosis in neuronal cells infected with Chlamydophila (Chlamydia) pneumoniae. BMC Neurosci. 2008;9:13. PubMed.
Further Reading
Papers
- Berthet A, Margolis EB, Zhang J, Hsieh I, Zhang J, Hnasko TS, Ahmad J, Edwards RH, Sesaki H, Huang EJ, Nakamura K. Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J Neurosci. 2014 Oct 22;34(43):14304-17. PubMed.
News
- Think Locally—Dendritic Mitochondria Required for Learning and Memory?
- Pink Fission—Serving Up a Rationale for Parkinson Disease?
- Form or Function: How Does Pink1 Deficiency Mar Mitochondria?
- Could Too Much Tau Be a Stretch for Mitochondria?
- Parkinsonism-linked Protein Binds Parkin and Pink1, Drives Mitophagy
- Mitochondrial Mutation Linked to Syndrome With ALS-FTD Features
- Neurons and Cancer Cells Share Survival Tactics
- NO Kidding? Mitochondria Fission Protein Linked to Neurodegeneration
- Prague: Aβ Rehabilitated as an Antimicrobial Protein?
Primary Papers
- Szelechowski M, Bétourné A, Monnet Y, Ferré CA, Thouard A, Foret C, Peyrin JM, Hunot S, Gonzalez-Dunia D. A viral peptide that targets mitochondria protects against neuronal degeneration in models of Parkinson's disease. Nat Commun. 2014 Oct 21;5:5181. PubMed.
- Lövheim H, Gilthorpe J, Johansson A, Eriksson S, Hallmans G, Elgh F. Herpes simplex infection and the risk of Alzheimer's disease-A nested case-control study. Alzheimers Dement. 2014 Oct 7; PubMed.
- Lövheim H, Gilthorpe J, Adolfsson R, Nilsson LG, Elgh F. Reactivated herpes simplex infection increases the risk of Alzheimer's disease. Alzheimers Dement. 2015 Jun;11(6):593-9. Epub 2014 Jul 17 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
National Institute on Aging
The paper by Szelechowski et al. is very interesting and another example of how studying viruses can lead to insights into broader biological processes. I find it intriguing that viruses that are dormant in neurons might protect their cellular hosts by limiting damage that could normally lead to neurodegeneration. If this is a general phenomenon, we might be able to find other ways to protect neurons against the normal low levels of stress we expect them to be under throughout their lifespan. In this particular paper, the axonal protection was particularly impressive. Such protection might be helpful, as the authors state, for those disorders that we currently view as being related to mitochondrial function in axons, especially where the damage is thought to be cell autonomous.
The one area of caution for me is that the MPTP model has an uncertain relationship to PD pathogenesis. While it is clear that people with MPTP exposure get PD-like symptoms, and some forms of parkinsonism are related to loss of function in mitochondrial processes, it is less clear if MPTP is a good model for all aspects of PD. MPTP provides a great way to lesion the intact nervous system and is reported to damage axons, but it would be very interesting to see if protein-X is active in other models. For example, it is reported that axonal damage occurs in rats where α-synuclein is overexpressed (Lundblad et al., 2012). This might make an interesting second model in which to try this neuroprotective strategy. A protein that is also protective in α-synuclein models might be valuable to pursue further as a therapeutic strategy for PD and related disorders.
References:
Lundblad M, Decressac M, Mattsson B, Björklund A. Impaired neurotransmission caused by overexpression of α-synuclein in nigral dopamine neurons. Proc Natl Acad Sci U S A. 2012 Feb 28;109(9):3213-9. PubMed.
View all comments by Mark CooksonUniversities of Manchester and Oxford
I agree with Brian Balin's comment on Madolyn Bowman Rogers' article about the intriguing effect of the Borna disease virus X protein, and would add only that it is in the interest of Borna and other viruses not to kill cells as in doing so, the virus too would succumb. In the case of HSV1, entering a state of latency in the host is a means of survival, providing a permanent reservoir of virus, despite frequent reactivations leading to productive infection and cell death.
On the theme of a viral role in AD, It is worth stressing that there are now some 80 papers by several groups (including those by Lovheim and colleagues), including 40 from mine, that directly or indirectly support the role of HSV1 in AD. These are striking in their use of remarkably diverse approaches—epidemiological, virological, genetic, and cell biological. My recent review (Itzhaki, 2014) summarizes them and explains certain relevant concepts in the hope that they will be better understood by those in the AD field who oppose a viral role. In fact, opposition to a role of HSV1 in AD has been expressed in a mere two publications, 11 and 13 years ago, in which the authors described their inability to detect HSV1 DNA in elderly and AD brains; however, six papers by groups other than mine have detected HSV1 DNA in such brains.
As for treating AD with antivirals with the aim of reducing disease progression, studies on three antivirals that act by different mechanisms have recently been published by my group (Wozniak et al., 2011; Wozniak and Itzhaki, 2013; Wozniak et al. 2013). Each antiviral was found to reduce substantially the accumulation of Aβ and phospho-tau that occurs in HSV1-infected cell cultures. The antiviral most likely to be used for therapy, acyclovir (or in practice its biodrug, valacyclovir), is very safe, and has the great advantage over many other treatments for AD that it targets only infected cells. Also, it should reduce or stop all viral damage, including the accumulation of Aβ and p-tau, irrespective of whether they are actual causes of the disease. There have been 413 AD trials between 2002 and 2012, with a failure rate of 99.6 percent (Cummings et al., 2014). Surely the time is now ripe for a clinical trial of an antiviral to combat HSV1 in the disease.
References:
Itzhaki RF. Herpes simplex virus type 1 and Alzheimer's disease: increasing evidence for a major role of the virus. Front Aging Neurosci. 2014;6:202. Epub 2014 Aug 11 PubMed.
Wozniak MA, Frost AL, Preston CM, Itzhaki RF. Antivirals reduce the formation of key Alzheimer's disease molecules in cell cultures acutely infected with herpes simplex virus type 1. PLoS One. 2011;6(10):e25152. PubMed.
Wozniak MA, Itzhaki RF. Intravenous immunoglobulin reduces beta amyloid and abnormal tau formation caused by herpes simplex virus type 1. J Neuroimmunol. 2013 Apr 15;257(1-2):7-12. PubMed.
Wozniak MA, Frost AL, Itzhaki RF. The helicase-primase inhibitor BAY 57-1293 reduces the Alzheimer's disease-related molecules induced by herpes simplex virus type 1. Antiviral Res. 2013 Sep;99(3):401-4. PubMed.
Cummings JL, Morstorf T, Zhong K. Alzheimer's disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6(4):37. Epub 2014 Jul 3 PubMed.
Make a Comment
To make a comment you must login or register.