Memory Retrieval, Not Storage, Hinders Mouse Models of Alzheimer’s
Quick Links
In early Alzheimer’s disease, people have trouble remembering what they did days, or even hours, prior. Is this because they lose the ability to store new memories, or are memories formed, but not properly retrieved? A study published online March 16 in Nature points to the latter. Susumu Tonegawa and colleagues at MIT report that young AD mice code fear memories just fine, but lack enough dendritic spines to recall them. When the researchers used optogenetics to beef up the number of synapses, the mice remembered as well as controls did. Researchers praised the work, suggesting it raises hopes of improving memory in people with AD. “We knew that Alzheimer’s was associated with a loss of spines, but we did not know how that affected cognition,” noted Alcino Silva, University of California, Los Angeles, who was not involved in the work. The results hint that spine loss impairs retrieval, not encoding, leaving open the possibility that lost memories could be restored in people, he said.
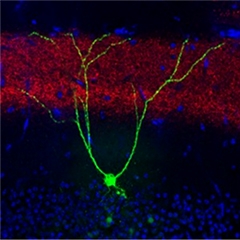
Memory cell.
A memory engram neuron in the dentate gyrus (green) receives inputs from the entorhinal cortex (red). Other neurons are labeled blue. [Courtesy of Dheeraj Roy.]
Previous studies using episodic memory tests suggested that people with Alzheimer’s have trouble forming new memories (for a review, see Weintraub et al., 2012). However, since tasks used to reach this conclusion rely on recall, it has been hard to rule out retrieval problems. Tonegawa’s group has developed gene expression and optogenetics techniques that can label and reactivate neurons used to form a particular memory, which are also known as engram cells (Liu et al., 2012). They previously reported that amnesic mice that apparently failed to store a conditioned fear response actually displayed it when engram cells were stimulated (Ryan et al., 2015). This suggested that the mice formed memories, but had trouble retrieving them. In the current study, first author Dheeraj Roy wondered if mice that model Alzheimer’s disease had similar recall problems.
To find out, he use contextual fear conditioning to teach mice to associate a cage with a mild foot shock. Then, to see if he could improve recall, he implanted and activated a light-sensitive ion channel in cells that stored that memory. He singled out these cells by expressing two adeno-associated viral vectors in the mouse hippocampus. One contained the gene for the tetracycline transactivator (tTA) driven by the c-fos promoter. The other expressed the light-activated channelrhodopsin 2 (ChR2) gene driven by a tTA response element. Since c-fos is essential for new memories, any memory engram cell would also make tTA. It would then express ChR2, unless the mice were fed doxycline, which suppresses tTA activity. Because ChR2 remains in the cell membrane for about three weeks, the scientists could come back during that time to check if memories were still encoded by zapping neurons with light from an optic fiber inserted into the dentate gyrus. If the memory of the foot shock was still there, the mice would freeze in response to the light, just as they would have in the fear-conditioning cage. They coupled ChR2 to enhanced yellow fluorescent protein to easily identify engram cells (see image above).
Roy tried this in seven-month-old APPswe/PSEN1dE9 mice, which had not yet accumulated amyloid plaques. These mice displayed normal short-term memory, freezing in the cage an hour after training. However, 24 hours after contextual fear conditioning, the mice froze much less frequently than controls, suggesting impairment in long-term memory. This is the equivalent of what a person might recall after five to 10 years, said Roy. Surprisingly, if he activated the engram cells a day later with light, the mice froze, but only if not eating doxycline in their chow. The scientists interpreted this to mean the memories were there, but that the mice just needed help recalling them. The researchers saw the same effect in 3xTG models infected with the double AAV engram system.
Upon closer inspection of the yellow fluorescent engram cells, Roy noticed that their dendritic spines weren’t as dense as those from control mice (see image below). Could this explain the retrieval problem? Since loss of synaptic density correlates with memory deficits in people with AD, Roy considered this a possibility (Terry et al., 1991).
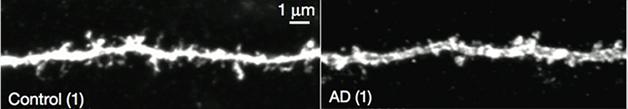
Seven-month-old APP/PS mice lose dendritic spines. [Courtesy of Roy et al., Nature.]
To find out, he tried boosting the number of dendritic spines on engram neurons by using long-term potentiation, which causes spines to sprout and enlarge. He injected an AAV vector encoding a faster-acting variant of ChR2, called oChIEF, again driven by the c-fos/tTa, into neurons of the entorhinal cortex (EC), upstream of the dentate gyrus memory cells. This allowed him to activate the EC engram cells with light at the high frequencies (100Hz) needed to induce long-term potentiation (LTP), strengthening their connections to dentate gyrus neurons. This raised the density of spines on dentate gyrus cells to control levels and restored context-specific fear memories for the next six days. Stimulating all the EC inputs to the dentate gyrus did not restore memories, however, implying the method depends on activation of specific engram cells. Likewise, if the researchers expressed diphtheria toxin receptor instead of the channelrhodopsin in the engram cells, then diphtheria toxin wiped out these cells and no memories were rescued.
Together, the results suggest that in early disease stages, loss of dendritic spines keeps mice—and perhaps humans—from recalling memories, said Roy. The work suggests that lost memories could be restored in people, too, but it’s not yet clear how, he said. Since stimulating dentate gyrus cells non-specifically did not restore memories, tools for generally activating neurons, such as deep-brain stimulation, may be too blunt, he said. Also, any therapies based on these data will likely work only early in disease, before neurons and their connections start to deteriorate.
Roy next plans to test other types of memory in mouse models and probe the specific cellular changes that strengthen synapses in LTP.
“This paper strongly supports the view that β-amyloid causes memory deficits by weakening synaptic strength,” wrote Sadegh Nabavi of Aarhus University, Denmark, to Alzforum. He proposed clearing β-amyloid from the circulation of mice after they formed memories, to see if it improved retrieval, as LTP did. This could suggest whether amyloid-clearing drugs would help restore memories.
“The potential to rescue long-term memory in dementia is exciting,” wrote Prerana Shrestha and Eric Klann of New York University in an accompanying News and Views. “In the future, Roy and colleagues’ findings might help to guide engram-based strategies that rescue memory deficits in patients with early stage Alzheimer’s disease.” However, they pointed out that some non-specific cells were probably labeled in the 24 hours mice were off doxycycline, so future methods might exploit a smaller time window.
Larry Squire, University of California, San Diego, said it was an interesting paper, and pointed out that the results might still be interpreted to indicate a deficit in encoding. Memories may be more weakly represented in AD mice, and require a strong manipulation—in this case, optogenetic stimulation—to elicit them, he said.—Gwyneth Dickey Zakaib
References
Research Models Citations
Paper Citations
- Weintraub S, Wicklund AH, Salmon DP. The neuropsychological profile of Alzheimer disease. Cold Spring Harb Perspect Med. 2012 Apr;2(4):a006171. PubMed.
- Liu X, Ramirez S, Pang PT, Puryear CB, Govindarajan A, Deisseroth K, Tonegawa S. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature. 2012 Apr 19;484(7394):381-5. PubMed.
- Ryan TJ, Roy DS, Pignatelli M, Arons A, Tonegawa S. Memory. Engram cells retain memory under retrograde amnesia. Science. 2015 May 29;348(6238):1007-13. Epub 2015 May 28 PubMed.
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991 Oct;30(4):572-80. PubMed.
Further Reading
Primary Papers
- Roy DS, Arons A, Mitchell TI, Pignatelli M, Ryan TJ, Tonegawa S. Memory retrieval by activating engram cells in mouse models of early Alzheimer's disease. Nature. 2016 Mar 24;531(7595):508-12. Epub 2016 Mar 16 PubMed.
- Shrestha P, Klann E. Alzheimer's disease: Lost memories found. Nature. 2016 Mar 24;531(7595):450-1. Epub 2016 Mar 16 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
RIKEN Center for Brain Science
First of all, I would like to praise the hard work of Roy and other authors. By definition, however, early AD is the clinical condition that arises after MCI, where tauopathy and neurodegeneration had already started. So the models that the authors used in this study are not models of early AD. Another model used, the 3XAD mice, are not even a model of AD because the tau mutation used causes FTDP-17, not AD.
Bateman and colleagues, 2012, showed that a minor cognitive decline starts after Aβ deposition approximately 10 years before clinical AD onset in humans. So, Roy's finding may account for this preclinical minor cognitive decline, but contextual fear conditioning would be very hard even for humans under these conditions to forget. Twenty-four hours is equally 24 hours for mice and for humans, in my view.
Make a Comment
To make a comment you must login or register.