A Molecular "Team Effort" in Synaptic Plasticity
Quick Links
One reason to study how brain systems develop is to understand how developmental and neurodegenerative diseases sabotage them. As an example, Alzheimer researchers might want to pay attention to a new study about synaptic plasticity during early development that coheres with an emerging view about the loss of cognitive function in Alzheimer disease (AD). The study appearing in the May 21 Nature Neuroscience focuses on several key molecules (i.e., NMDAR, BDNF, and PSD-95) that are involved in normal synaptic formation early in life but are also affected in AD. The interactions of these molecules are not well understood, but the authors piece them together into a direct linear pathway with a positive feedback loop, one that they suggest may help ingrain memories.
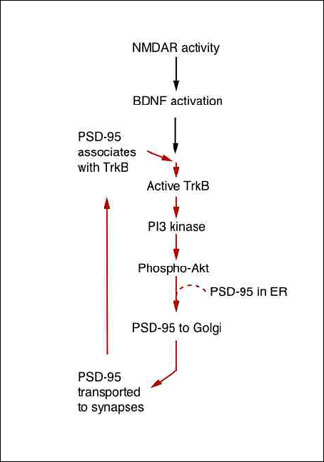
Suggested Positive Feedback Loop of NMDA/BDNF/PSD-95 Pathway
Both NMDAR stimulation and BDNF exposure initiate a linear pathway that transports new PSD-95 to the synapses, where it serves as a scaffold for glutamate receptors, BDNF receptors, and other signaling molecules. Furthermore, local BDNF stimulation drives PSD-95 trafficking to synapses throughout the neuron, indicating rapid, dendrite-wide sensitization for synaptic potentiation. Image credit: Akira Yoshii and Martha Constantine-Paton
The model of this new pathway’s role during development, learning, and memory ties in with the recent recognition that in AD, β amyloid (Aβ) accumulation results in the loss of synapses, which is a significant factor in the cognitive decline and memory loss, comments senior author Martha Constantine-Paton, at the McGovern Institute for Brain Research at MIT. “We hope this study might call attention to this pathway’s potential relevance for Alzheimer’s research.”
Constantine-Paton and first author Akira Yoshii focus on eye opening. This is a major developmental event in rodents. It happens after birth and is followed by rapid increases in synapse strength and visual circuit refinement in response to the onset of visual stimulation. Previously, the authors had linked this increase in synaptic strength to the post-synaptic density protein PSD-95, which rushes to the synapses on the neuron’s dendritic spines within hours of eye opening. PSD-95 is a scaffold that anchors NMDA and AMPA receptors for glutamate. Glutamate triggers the neuron’s electrical activity during development, learning, and memory. PSD-95 is known to anchor many other signaling and structural molecules, but the authors were surprised to discover that it also holds the TrkB receptor for brain-derived neurotrophic factor (BDNF), an essential growth factor for synaptic plasticity, neuronal survival, and memory formation.
In the current work, the researchers explored the relationship between PSD-95 and BDNF. They found that stimulating the NMDA receptor activates BDNF, which initiates the following cascade. Namely, BDNF triggers a signaling pathway involving another well-studied duo, PI3 kinase/AKT. That pathway leads to more PSD-95, and with it more receptors for BDNF, to accumulate rapidly at the synapse. As a result, the synapse becomes more responsive to BDNF, which sends more PSD-95 to the synapse in a positive feedback loop.
Yoshii and Constantine-Paton speculate that this pathway may intersect with the downward spiral of synaptic loss on dendritic spines in Alzheimer disease. They point to the growing consensus that the production and accumulation of the Aβ peptide is central to the pathogenesis of AD, and that Aβ exacts its toll by targeting the synapse itself and its functions. One recent study showed that Aβ removes AMPA receptors at synapses, which induces synaptic depression and eventual loss of dendritic spines and NMDA responses (Hsieh et al., 2006). A different study showed that certain kinds of Aβ oligomer cause dendritic spine loss through the NMDAR pathway (Shankar et al., 2007). Likewise, Tong et al., 2004) reported that in cultured neurons, Aβ antagonizes the BDNF-induced phosphorylation and activation of AKT, which mediates the newly discovered pathway induced by BDNF.
“Although our study focuses on formation of young synapses, we can learn a lot from developmental neurobiology about the mature or even aging brain,” comments Yoshii. “It will be interesting to see if the spine loss induced by Aβ perturbs the synaptic delivery of PSD-95 through this newly discovered pathway.”—Cathryn Delude
Cathryn Delude is a freelance writer based in Andover, Massachusetts.
References
Paper Citations
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006 Dec 7;52(5):831-43. PubMed.
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007 Mar 14;27(11):2866-75. PubMed.
- Tong L, Balazs R, Thornton PL, Cotman CW. Beta-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J Neurosci. 2004 Jul 28;24(30):6799-809. PubMed.
Further Reading
Primary Papers
- Yoshii A, Constantine-Paton M. BDNF induces transport of PSD-95 to dendrites through PI3K-AKT signaling after NMDA receptor activation. Nat Neurosci. 2007 Jun;10(6):702-11. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.