More Mitochondrial Mayhem in ALS Motor Neurons, Muscles?
Quick Links
Much is amiss with mitochondria during the pathology of amyotrophic lateral sclerosis. Two new papers report on how the cell’s powerhouse organelle is thrown off balance by mutant proteins linked to the disease. In the January 18 Journal of Neuroscience, researchers from Children’s Hospital of Philadelphia, Pennsylvania, describe how blocking AMP activated protein kinase—normally a beneficial modulator of cellular metabolism—protects motor neurons in cell and animal models of the disease. Going beyond the motor neuron and into the muscle, a team based at the University of Alabama School of Medicine in Birmingham report, in the January 19 Developmental Cell online, that a mutation associated with ALS disrupts normal mitochondrial architecture in worms and flies.
Together, the two works provide more fodder for the theory that ALS involves major energetic dysfunction, commented Jordi Magrané of the Weill Medical College of Cornell University in New York City. Magrané, who was not involved in the current studies, has described abnormal mitochondrial transport in motor neurons modeling ALS (see ARF related news story on Magrané et al., 2012).
Neuronal Metabolism Out of Control
Robert Kalb, senior author on the Journal of Neuroscience paper, was inspired to examine energy regulation in ALS by previous work linking defective metabolism to the disease in mice (Dupuis et al., 2004). He recruited graduate student and first author Ria Lim to examine the role of AMP activated protein kinase (AMPK), a master regulator of cellular metabolism, in the disease. AMPK is activated when the ratio of ATP:AMP drops too low. For example, during exercise, AMPK turns on in muscle tissue and helps the cell burn the extra fuel it needs. The kinase, Kalb and Lim suspected, would mediate the cell’s response to abnormal ATP levels in ALS as well.
The researchers cultured neurons from embryonic rat spinal cords and expressed the gene for human superoxide dismutase 1 (SOD1) in the cells. Mutant SOD1, which causes ALS, is known to bind the anti-apoptotic protein Bcl-2 in spinal cord mitochondria (see ARF related news story on Pasinelli et al., 2004) and block mitochondrial pores (see ARF related news story on Israelson et al., 2010). In Lim’s assay, the G37R mutant SOD1 killed the majority of cultured neurons within three days, while cells expressing the wild-type dismutase survived. The mSOD1 cells also exhibited depressed respiration. While it is not easy to directly measure the ATP:AMP ratio in cultures, Kalb said, the reduced oxygen use that Lim measured indicated reduced metabolism. As an energy sensor, AMPK should respond to this deficiency, with a conformational change that makes it more likely to be phosphorylated by a handful of upstream kinases. Sure enough, Lim observed 50 percent more active, phosphorylated AMPK in cells making mSOD1 compared to wild-type neurons.
In normal cells, AMPK keeps ATP levels steady by boosting uptake of fatty acids and glucose, while downregulating other activities like protein translation that use up fuel. Would this activity protect the sickened neuron cultures expressing mSOD1? To find out, Lim treated the cells with an AMPK antagonist, Compound C, or an activator, AICAR. Compound C kept the neurons alive, but AICAR did not, indicating AMPK activation is bad news for motor neurons with mSOD1, and that dampening its function is protective.
Lim and Kalb took their experiments in vivo with C. elegans that express human G85R SOD1 in their neurons. These nematodes are impaired in movement (Wang et al., 2009). Lim crossed these worms with a strain lacking aak-2, a subunit of AMPK. The double mutants didn’t move quite like wild-type worms, but they crawled and swam more than twice as quickly as mSOD1 single mutant worms. Similarly, aak-2 deletion alleviated some of the movement problems of worms expressing mutant TDP-43, another ALS-linked gene.
Although the complete pathway from mutant SOD1 expression to metabolic defects remains unknown, “we think that cells are sensing a bioenergetic defect, they are engaging [AMPK] to correct that defect, and the systems they engage are, in fact, detrimental,” Kalb concluded. Next, he intends to examine which body tissues are most strongly affected by the energy-managing dysfunction, and look for potential drug targets. Neurons, he speculated, could be affected because they expend loads of ATP to maintain their ionic gradients.
Muscle Mitochondria Mislocalized
While motor neurons are the major focus of ALS research, muscle garners more attention lately (see ARF related news story on Palma et al., 2011). In the Developmental Cell paper, first author Sung Min Han and senior author Michael Miller, both at the University of Alabama, reported how motor neurons affect mitochondria in muscles.
Han and Miller focused on VAPB, another ALS gene (Nishimura et al., 2004). Miller noticed that VAPB contains a motif called a major sperm protein (MSP) domain, named for the factor that worms' sperm secrete to cause oocyte maturation (Miller et al., 2001). He suspected that the MSP domain of VAPB could also be secreted, a fact which he and Han confirmed in collaboration with Hiroshi Tsuda, then a postdoctoral researcher in the laboratory of Hugo Bellen at Baylor College of Medicine in Houston, and now at McGill University in Montréal, Canada. They also found that the ALS-associated P58S mutation in the MSP domain prevents this secretion (see ARF related news story on Tsuda et al., 2008).
In the current work, Han and Miller, Tsuda and Bellen again collaborated to elucidate the importance of VAPB MSP secretion. First, they examined Drosophila lacking the VAPB homolog dvap. Studying flight muscles, they found that mitochondria neatly line up with actin in between the myosin-containing striations in wild-type flies. But in the dvap-null insects, the mitochondria were deformed; most were puny, while some were swollen. Similarly, in worms missing the VAPB homolog vpr-1, muscle mitochondria failed to form their normal organized lines and instead sprawled across the cytoplasm in highly interconnected networks. The worms’ mitochondria also took up only small amounts of the marker MitoTracker, which depends on a healthy membrane potential for its uptake, indicating poor mitochondrial function. The worms themselves were “lethargic,” Miller said, further confirming their metabolic syndrome.
If lack of VAPB, or its homologs, causes problems, then replacing it might fix the phenotype. When the researchers provided dvap to the muscle of dvap-null flies, it did not rescue the muscle defects; however, putting dvap specifically in the neurons did. The same was true in the nematodes. “These VAP proteins are essentially like secreted motor neuron factors that regulate energy production in muscle,” Miller concluded. Given how the ALS-linked mutation disables VAPB MSP secretion, “that provides some compelling evidence that the secreted form is crucial for the pathogenesis,” he added. VAPB was also recently reported to have a key cell-autonomous role in maintaining proper calcium uptake by mitochondria (De Vos et al., 2011).
In further experiments, the team identified SAX-3 Robo, and CLR-1 Lar receptors as the key muscle receptors for the VAPB MSP domain. Signaling pathways downstream of these receptors regulate the actin cytoskeleton, which controls mitochondrial localization (see figure below). The same receptors are expressed in neurons, so the VAPB MSP domain might influence neural mitochondrial arrangements as well, the authors note.
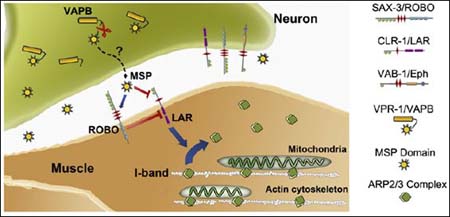
Motor Neuron VAPB Fragment Energizes Muscle
Motor neurons cleave and secrete the VAPB MSP subunit. ROBO and LAR receptors on muscle cells pick up the MSP, which alters downstream signaling to promote integration of mitochondria into I-bands, the actin-rich striations where they belong. ALS-linked VAPB mutants fail to release MSP, linking these mutations to muscle dysfunction in disease. Image credit: Cell Press
“One major unresolved question is how the VAP-MSP fragment reaches the extracellular space. Another question is whether a similar mechanism operates in vertebrates and, in particular, in mammals,” noted Dick Jaarsma of the University Medical Center Rotterdam, The Netherlands, in an e-mail to ARF (see full comment below). “The model of Han et al. predicts that the functional deficits resulting from VAPB deficiency in motor neurons occur in the muscle fibers. However, electromyography and muscle biopsy point to a neurogenic basis of the disease…implying functional loss of motor neurons or their axons (rather than muscle fibers) as the prime event in the disease,” Jaarsma continued. Miller noted that in ALS, “You see a lot wrong with the muscle, too,” and suggested there is a renewed appreciation for the role of muscle in the ALS disease process.
Magrané was excited to see that the VAPB, like SOD1, causes mitochondrial mislocalizations. So do mutations in the ALS gene TDP-43, as the Kalb team and others have observed (see ARF related news story on Xu et al., 2010). “It looks like [ALS pathology] is all about positioning; it is all about having mitochondria where they are supposed to be,” Magrané said.—Amber Dance
References
News Citations
- Mitochondria Stumble Their Way Along Axons in ALS Model
- Motoneuron Mitochondria: Preferred Destination For Mutant SOD1
- New Theory for Some ALS Cases—SOD1 Plugs Cell Power Plants
- ALS Muscle Matters: Biopsy and Frog Egg Models Probe Pathology
- Less VAPid Now: Role for ALS Protein Gets Substance
- Paper Alert: Malformed Mitochondria in the Latest TDP-43 Mouse
Paper Citations
- Magrané J, Sahawneh MA, Przedborski S, Estévez ÁG, Manfredi G. Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J Neurosci. 2012 Jan 4;32(1):229-42. PubMed.
- Dupuis L, Oudart H, René F, Gonzalez de Aguilar JL, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci U S A. 2004 Jul 27;101(30):11159-64. PubMed.
- Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004 Jul 8;43(1):19-30. PubMed.
- Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V, Cleveland DW. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010 Aug 26;67(4):575-87. PubMed.
- Wang J, Farr GW, Hall DH, Li F, Furtak K, Dreier L, Horwich AL. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009 Jan;5(1):e1000350. PubMed.
- Palma E, Inghilleri M, Conti L, Deflorio C, Frasca V, Manteca A, Pichiorri F, Roseti C, Torchia G, Limatola C, Grassi F, Miledi R. Physiological characterization of human muscle acetylcholine receptors from ALS patients. Proc Natl Acad Sci U S A. 2011 Dec 13;108(50):20184-8. PubMed.
- Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, Kok F, Oliveira JR, Gillingwater T, Webb J, Skehel P, Zatz M. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004 Nov;75(5):822-31. PubMed.
- Miller MA, Nguyen VQ, Lee MH, Kosinski M, Schedl T, Caprioli RM, Greenstein D. A sperm cytoskeletal protein that signals oocyte meiotic maturation and ovulation. Science. 2001 Mar 16;291(5511):2144-7. PubMed.
- Tsuda H, Han SM, Yang Y, Tong C, Lin YQ, Mohan K, Haueter C, Zoghbi A, Harati Y, Kwan J, Miller MA, Bellen HJ. The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell. 2008 Jun 13;133(6):963-77. PubMed.
- De Vos KJ, Mórotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, Warley A, Shaw CE, Miller CC. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet. 2011 Dec 13; PubMed.
- Xu YF, Gendron TF, Zhang YJ, Lin WL, D'Alton S, Sheng H, Casey MC, Tong J, Knight J, Yu X, Rademakers R, Boylan K, Hutton M, McGowan E, Dickson DW, Lewis J, Petrucelli L. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010 Aug 11;30(32):10851-9. PubMed.
Further Reading
Papers
- Karbowski M, Neutzner A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol. 2012 Feb;123(2):157-71. PubMed.
- Martin LJ. Mitochondrial pathobiology in ALS. J Bioenerg Biomembr. 2011 Dec;43(6):569-79. PubMed.
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CC, Grierson AJ. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007 Nov 15;16(22):2720-8. Epub 2007 Aug 28 PubMed.
- Fischer LR, Igoudjil A, Magrané J, Li Y, Hansen JM, Manfredi G, Glass JD. SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain. 2011 Jan;134(Pt 1):196-209. PubMed.
- Li Q, Vande Velde C, Israelson A, Xie J, Bailey AO, Dong MQ, Chun SJ, Roy T, Winer L, Yates JR, Capaldi RA, Cleveland DW, Miller TM. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc Natl Acad Sci U S A. 2010 Dec 7;107(49):21146-51. PubMed.
- Chiang PM, Ling J, Jeong YH, Price DL, Aja SM, Wong PC. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci U S A. 2010 Sep 14;107(37):16320-4. PubMed.
- Kawamata H, Manfredi G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech Ageing Dev. 2010 Jul-Aug;131(7-8):517-26. PubMed.
- Magrané J, Hervias I, Henning MS, Damiano M, Kawamata H, Manfredi G. Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum Mol Genet. 2009 Dec 1;18(23):4552-64. PubMed.
News
- Motoneuron Mitochondria: Preferred Destination For Mutant SOD1
- Less VAPid Now: Role for ALS Protein Gets Substance
- Motors and Muscles—Pacing ALS Progression
- In ALS, “Good” Mitochondrial Protein Turns Partner in Crime
- Paper Alert: Malformed Mitochondria in the Latest TDP-43 Mouse
- New Theory for Some ALS Cases—SOD1 Plugs Cell Power Plants
- San Diego: Mutant SOD1 Bumps Mitochondrial Current Up—Or Down?
- ALS: Mutant Enzyme Does Not Hang Around, But Does Find Mitochondria
- Mitochondria Stumble Their Way Along Axons in ALS Model
- Mitochondrial Chromosome Disintegrates in ALS Motor Neurons
- Being Pleasantly Plump: Way to Live Longest with ALS?
- Mutant Meddling in Mitochondria Partly Mimics ALS Pathology
- ALS Muscle Matters: Biopsy and Frog Egg Models Probe Pathology
Primary Papers
- Lim MA, Selak MA, Xiang Z, Krainc D, Neve RL, Kraemer BC, Watts JL, Kalb RG. Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J Neurosci. 2012 Jan 18;32(3):1123-41. PubMed.
- Han SM, Tsuda H, Yang Y, Vibbert J, Cottee P, Lee SJ, Winek J, Haueter C, Bellen HJ, Miller MA. Secreted VAPB/ALS8 major sperm protein domains modulate mitochondrial localization and morphology via growth cone guidance receptors. Dev Cell. 2012 Feb 14;22(2):348-62. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
�
I think this is a great paper that unravels a novel role for VAMP/synaptobrevin-associated protein B (VAPB) with great precision. The paper again illustrates the power of Drosophila and C. elegans as animal models for identifying functional roles of proteins and illuminating potential disease pathways.
The questions raised by the P56S-VAPB mutation are essentially the same as those raised by mutations in genes linked to other ALS forms, as well as to other neurodegenerative disorders: What is the function of the mutated protein? Does the disease result from a loss of its normal function or a gain of toxic activity? What is the role of protein aggregation? What determines the delayed onset of disease, and to what extent do disease pathways overlap with those in other ALS forms? And, most importantly, how to stop or prevent disease?
Several lines of evidence indicate that mutant VAPB, at least in part, may operate in a dominant-negative way by recruiting wild-type VAPB to inclusions. the paper by Han et al. provides one potential mechanism by which loss of VAPB function in motor neurons results in reduced performance of skeletal muscle.
This work follows a provocative study by the same groups published in Cell in 2008, indicating that a fragment of the Drosophila VAPB homologue containing the major sperm protein (MSP) domain may act as a paracrine factor released by motor neurons at the neuromuscular junctions. Now, Han et al. show that loss of VAPB in motor neurons in both Drosophila and C. elegans results in structural and functional mitochondrial abnormalities in the target muscles. Importantly, the same effect was also observed after mutant VAP overexpression in the motor neurons, supporting the dominant-negative mode of action of mutant VAP.
In an elegant series of experiments, the authors further show that the MSP domain of VAPB binds to a Robo and a Lar-receptor to control mitochondrial localization via the regulation of the actin skeleton. As indicated in the conclusion scheme by the authors (Fig. 8C), one major unresolved question is how the VAP-MSP fragment reaches the extracellular space. Another question is whether a similar mechanism operates in vertebrates and, in particular, in mammals. This question awaits the analysis of VAPB-knockout mice.
Another problem is that the authors’ model predicts that the functional defects resulting from VAPB deficiency in motor neurons occur in the muscle fibers. However, EMG and muscle biopsy (Marques et al., 2006) point to a neurogenic basis of the disease, consistent with the diagnosis of ALS, and implying functional loss and degeneration of motor neurons or their axons (rather than muscle fibers) as the prime event in the disease. Nevertheless several mechanisms can be envisaged by which loss of motor neuron VAPB contributes to muscle weakness not only in VAPB-ALS, but also in other ALS forms.
Remarkably, a recent study has suggested a mechanism by which mutant VAPB may cause mitochondrial abnormalities in a cell-autonomous way in motor neurons (De Vos et al., 2011). This study shows that VAPB may interact with the mitochondrial protein PTPIP51 to regulate its interaction with the endoplasmic reticulum and calcium homeostasis. According to this study, mutant VAPB disturbs this interaction and alters calcium homeostasis. In regard to that study, I would like to point out that we do not see ultrastructural mitochondrial abnormalities in motor neurons of our transgenic mice that overexpress mutant VAPB. The mice develop numerous VAPB "aggregates" in motor neurons, but the aggregates seem to be rather harmless, which is consistent with data from another study with VAPB transgenic mice (Tudor et al., 2010).
References:
Marques VD, Barreira AA, Davis MB, Abou-Sleiman PM, Silva WA, Zago MA, Sobreira C, Fazan V, Marques W. Expanding the phenotypes of the Pro56Ser VAPB mutation: proximal SMA with dysautonomia. Muscle Nerve. 2006 Dec;34(6):731-9. PubMed.
De Vos KJ, Mórotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, Warley A, Shaw CE, Miller CC. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet. 2011 Dec 13; PubMed.
Tudor EL, Galtrey CM, Perkinton MS, Lau KF, De Vos KJ, Mitchell JC, Ackerley S, Hortobágyi T, Vámos E, Leigh PN, Klasen C, McLoughlin DM, Shaw CE, Miller CC. Amyotrophic lateral sclerosis mutant vesicle-associated membrane protein-associated protein-B transgenic mice develop TAR-DNA-binding protein-43 pathology. Neuroscience. 2010 May 19;167(3):774-85. Epub 2010 Feb 24 PubMed.
Make a Comment
To make a comment you must login or register.