New Role for PINK1 Offers Clues about Parkinson's Pathology
Quick Links
Researchers have zeroed in on a mechanism that may promote the earliest stages of mitochondrial decline in Parkinson’s disease (PD). Mutations that hinder the kinase activity of the mitochondria-associated protein PINK1 trigger the derailment of the electron transport chain that provides energy to the cell, according to work described in the March 20 ScienceXpress. Investigators led by Bart De Strooper of the University of Leuven in Belgium reported that defects in PINK1 compromised a protein complex in the respiratory chain. PINK1 previously has been described as promoting clearance of defective mitochondria, a process known as mitophagy. In addition, impairment of PINK1’s bioenergetic effects may underlie PD pathology, the researchers suggest.
“There is a big question in the field as to whether PINK1 is only working in the mitophagy pathway or has other functions,” Wade Harper of Harvard University in Cambridge, Massachusetts, wrote to Alzforum. “This paper would definitely suggest other functions.” Harper was not involved in the study.
Growing evidence supports the view that mitochondria malfunction in PD. This is exemplified by studies on two genes linked to familial forms of the disease, PINK1 (a kinase) and Parkin (an E3 ubiquitin ligase). Through complex interactions that researchers are still unraveling, the two proteins trigger the disposal of damaged mitochondria in the wake of cellular stress. This move spares cells from death. However, previous studies by De Strooper and others have indicated that cells lacking functional PINK1 also show signs of reduced activity of Complex I, the behemoth protein conglomerate that makes the first of a series of electron handoffs in the respiratory chain. The activity of the complex is crucial for the maintenance of the mitochondrial proton gradient that drives the production of adenosine triphosphate (ATP).
“By far the dominant hypothesis in the field is that PINK1 acts through Parkin in the mitophagy pathway,” De Strooper said. “There is a lot of truth in that, but you need to use rather drastic conditions to stimulate that mechanism.” For example, researchers need to treat cells with CCCP, a potent disruptor of mitochondrial membrane potential, to trigger mitophagy, which is when any defects in the PINK1/Parkin pathway surface. However, De Strooper’s lab reported in 2009 that cells from PINK1-deficient fruit flies and mice display Complex I defects in the absence of stress (see Morais et al., 2009). The evidence hinted that PINK1 plays separate roles in the mitophagy and energy pathways. However, whether the two paths are related or if they conspire to cause disease remains a mystery.
To understand how the kinase modulates Complex I, first author Vanessa Morais and colleagues tested whether PD-causing PINK1 mutations disrupt electron transport. In both fibroblasts and induced pluripotent stem cell (iPSC)-derived neurons from patients harboring such mutations, the researchers observed lower concentrations of ATP and a drop in mitochondrial membrane potential compared to controls.
Because Complex I is the primary gatekeeper of the electron transport chain, and PINK1 is a kinase, the researchers next employed a “phosphoproteome” approach to scan the complex for signs of phosphorylation deficiencies in the absence of PINK1. The investigators purified the complex from PINK1-deficient mouse mitochondria and used mass spectroscopy to take an inventory of phosphorylated amino acids. Only one residue—Serine 250 from a protein subunit called NdufA10—lacked a phosphate group found in normal mitochondria, suggesting this modification may be important for Complex I function. To test this, the researchers transfected the cells with NdufA10-S250D, which acts like a phosphorylated version of the subunit. This phosphomimetic rescued the deficits in mitochondrial membrane potential and ATP production in PINK1-deficient cells (see image below).
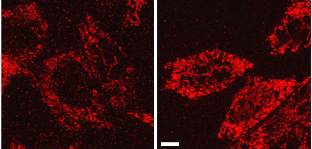
PINK1 Problems:
In the absence of Pink1, mitochondrial membrane potential plummets and the organelles have trouble producing ATP (red). A phosphomimetic of the NdufA10 subunit of Complex I of the respiratory chain restores membrane potential and ATP (right). [Image courtesy of Science/AAAS]
The phosphoproteome results do not prove that PINK1 directly phosphorylates NdufA10, This is the main limitation of the paper, said Harper, and De Strooper acknowledged that he cannot rule out the possibility of an intermediate kinase. However, Mark Cookson of the National Institute on Aging in Bethesda, Maryland, said it would be a tall order experimentally to prove PINK1 phosphorylates NdufA10 within the “bizarre” physiological setting of the space between the inner and outer mitochondrial membrane. Cookson, who was impressed with the phosphoproteomics approach, added that few protein kinases exist in the intermembrane space, so chances are good that PINK1 does the deed.
The researchers next used enzyme assays to measure the biochemical consequences of PINK1 deficiency in the respiratory chain. Complex I prepared from PINK1-deficient mouse fibroblasts failed to hand off electrons to coenzyme Q1 (CoQ1), the next protein down the line. Expression of the phosphomimetic reversed the defect, and the same was true in patient-derived fibroblasts expressing PINK1 mutations.
What role does NdufA10 phosphorylation play in mitochondria? To answer this, the investigators turned to Drosophila, because flies lacking PINK1 show more drastic neurological phenotypes than do mice. Neurons in PINK1-deficient flies fail to rapidly mobilize or replenish synaptic vesicles during high-frequency electrical stimulation, an energy-intensive action known to depend on Complex I activity and ATP production. The researchers rescued this defect by expressing the NdufA10 phosphomimetic. However, the phosphomimetic failed to rescue defects in mitophagy, suggesting that the process does not require NdufA10, and that PINK1 acts on mitophagy and the respiratory chain in distinct ways.
While dual roles could play out in parallel, the researchers suspect that in reality one precedes the other. “We are inclined to think that the mitophagy defect would be a consequence of the mitochondrial energy defect,” Morais said. Cookson agreed that this chain of events was likely, and that PINK1’s role of activating Complex I in the intermembrane space would conveniently position the protein to trigger the mitophagy pathway, should the need arise.—Jessica Shugart
References
Paper Citations
- Morais VA, Verstreken P, Roethig A, Smet J, Snellinx A, Vanbrabant M, Haddad D, Frezza C, Mandemakers W, Vogt-Weisenhorn D, Van Coster R, Wurst W, Scorrano L, De Strooper B. Parkinson's disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol Med. 2009 May;1(2):99-111. PubMed.
Further Reading
Papers
- Springer W, Kahle PJ. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy. 2011 Mar;7(3):266-78. PubMed.
- Song S, Jang S, Park J, Bang S, Choi S, Kwon KY, Zhuang X, Kim E, Chung J. Characterization of PINK1 (PTEN-induced putative kinase 1) mutations associated with Parkinson disease in mammalian cells and Drosophila. J Biol Chem. 2013 Feb 22;288(8):5660-72. PubMed.
Primary Papers
- Morais VA, Haddad D, Craessaerts K, De Bock PJ, Swerts J, Vilain S, Aerts L, Overbergh L, Grünewald A, Seibler P, Klein C, Gevaert K, Verstreken P, De Strooper B. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science. 2014 Apr 11;344(6180):203-7. Epub 2014 Mar 20 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.