Potent TREM2 Antibody Stirs Microglia to Prune Plaques in Mice
Quick Links
When scientists realized that certain Alzheimer's-linked variants in the microglial membrane receptor TREM2 were loss-of-function mutations, they were spurred to create drugs that make this receptor more active. The latest one? Researchers led by Zhiqiang An and Ningyan Zhang at the University of Texas Health Science Center, Houston, created Ab18, a tetravalent TREM2 antibody that bumps up receptor activation 100-fold. In the September 7 Science Translational Medicine, they reported that the antibody prompted TREM2 to cluster, boosting receptor signaling and encouraging microglia to migrate toward and ingest Aβ oligomers.
- New tetravalent antibody activates TREM2 100-fold.
- It prompts microglia to surround and engulf Aβ oligomers.
- Treated mice had far fewer plaques and tangles, better memories.
The researchers also created a bispecific version of Ab18 that better slips into the brain. When injected into a mouse model of amyloidosis, this antibody led to lower amyloid plaque and tau loads, denser synaptic fields, and better memory.
"The findings further validate immuno-neurology and Trem2 activators as exciting approaches to address Alzheimer’s disease and possibly other neurodegenerative disorders," wrote Arnon Rosenthal of Alector. “The consistency of these findings with those from other TREM2 agonists further strengthens the exciting possibility of therapeutically modulating microglial function,” wrote Christian Haass and Kai Schlepckow at the German Center for Neurodegenerative Diseases in Munich (see full comments below). Joseph Lewcock and Kathryn Monroe of Denali Therapeutics, South San Francisco, agreed. “It’s encouraging to see very similar results between Ab18 and our bispecific TREM2 antibody,” they told Alzforum. A Phase 1 study of Denali’s antibody, DNL919, began in Europe last month.
To begin his search for suitable antibodies, first author Peng Zhao screened a phage display library against the mouse TREM2 receptor expressed on human kidney cells. He identified four antibodies that bound TREM2 yet did not interfere with its interaction with natural ligands, including oligomeric Aβ and phospholipids. One of the four dose-dependently activated TREM2 and increased microglial uptake of fluorescently labeled Aβ oligomers.
Zhao increased the antibody’s potency by adding two more TREM2 binding sites to create a tetravalent version, dubbed Ab18. Compared to its bivalent predecessor, Ab18 increased TREM2 activation 100-fold, microglial migration to Aβ oligomers 300-fold, and microglial Aβ uptake 33-fold. “The unique architecture of this tetravalent antibody demonstrates how important valency is to TREM2 function,” Monroe said. She noted that this work extends previous findings that monovalent TREM2 antibodies do not induce signaling, bivalent ones do, and tetravalent antibodies are even more effective (Mar 2020 news).
What made Ab18 so potent? Efficient TREM2 signaling stems from clustering of the receptor on the microglial surface. The scientists wondered if Ab18’s higher valency encouraged this, so they turned to size exclusion chromatography and fluorescence microscopy to find out. Indeed, purified TREM2 formed larger complexes with Ab18 than with its bivalent counterpart. In mouse microglia treated with Ab18, a fluorescent dye labeling the antibody revealed puncta that the authors believed to be Ab18-TREM2 aggregates (see image below). Together, these results suggest that Ab18 induces clustering of TREM2 to dial up its signaling.

Clustering TREM2. In mouse neonatal microglia bathed in tetravalent Ab18 (right), a dye labeling the antibody (green) illuminated puncta, suggesting TREM2 clustering. In microglia given the bivalent version, few puncta formed (left). Nuclei are stained blue. [Courtesy of Zhao et al., Science Translational Medicine, 2022.]
To test their tetravalent TREM2 antibody in vivo, the researchers first increased its ability to enter the brain by attaching it to an antibody targeting the mouse transferrin receptor. Endothelial cells along the blood-brain barrier highly express TfR, allowing them to transport transferrin across the BBB. The Ab18-TfR easily slipped into the brain. Wild-type mice injected with this bispecific antibody had 10 times more of it in their brains than did animals injected with Ab18. Immunofluorescence of mouse brain slices showed Ab18-TfR outside the CD31-positive blood vessels, cozying up to Iba1-positive microglia in the parenchyma.
Next, the scientists repeated the cell experiments to confirm that, like Ab18, the bispecific version prompted TREM2 to cluster, activating it to improve microglial migration to, and phagocytosis of, Aβ oligomers.
How would the TREM2 antibody affect amyloid plaques? The researchers injected 5-month-old 5xFAD mice with Ab18-TfR weekly for 14 weeks, then collected brain tissue. Compared to mice injected with a control antibody, treated mice had three- and 10-fold fewer 6E10-positive amyloid plaques in their cortices and hippocampi, respectively (see image below). Plaques were smaller and more diffuse.
Brain tissue from treated mice had five times as many TREM2-expressing microglia encircling plaques as control tissue. These plaque-adjacent microglia expressed the phagocytosis marker CD68, suggesting that they were feasting on Aβ (see image below).
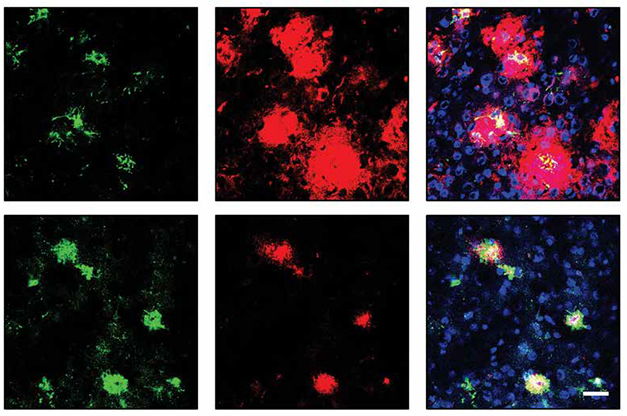
TREMing Down Plaques. Compared to controls (top), mice injected with Ab18-TfR (bottom) had smaller plaques (red) ringed by more microglia expressing the phagocytic marker CD68 (green). [Courtesy of Zhao et al., Science Translational Medicine, 2022.]
What about downstream Alzheimer's disease markers, such as tauopathy, axon damage, neuron loss? These mice don’t form neurofibrillary tangles; however, the scientists stained brain tissue for phosphorylated tau with the AT8 antibody, finding that Ab18-TfR halved total p-tau and p-tau co-localized with amyloid plaques. Treated mice had 60 percent less neurodegeneration as judged by total phosphorylated neurofilament heavy chain, pNF-H. They also had 80 percent fewer LAMP1-positive dystrophic neurites and 1.5-fold more NeuN-positive neurons. The authors concluded that Ab18-TfR lowered p-tau concentration and alleviated axon damage and neuron loss.
As for behavioral assays, treated mice spent half as much time in the open arm of an elevated plus maze and froze 50 percent more often during contextual fear tests, indicating that the antibody may have preserved fear of open spaces and memory.
Separately, the scientists performed the same optimization on another tetravalent TREM2 antibody, Ab2, which came out of a phage display screen to bind to human TREM2. In in vitro assays of human microglia and in mice injected with human microglia, Ab2-TfR elicited much the same response as Ab18. That work was reported August 3 in the journal MAbs. An told Alzforum that he is planning to develop Ab2-TfR for clinical use, and is partnering with a company, which he declined to name, for preclinical studies.
Other bivalent TREM2 antibody therapeutics are already in the works. Alector’s AL002, currently in a Phase 2 study, binds to and activates TREM2, which increases TREM2 signaling and induces microglial proliferation. Denali’s DNL919 includes a TfR antibody fragment within, rather than attached to, the TREM2 antibody’s Fc domain. This drug is poised to start a Phase 1 trial in the U.S. pending release of an FDA hold (see company press release). Lewcock said that Denali made the strategic decision to move a portion of the DNL919 Phase 1 trial to Europe, and that the company plans to bring interim clinical data to the FDA with the aim of beginning a U.S. trial.—Chelsea Weidman Burke
References
Mutation Interactive Images Citations
News Citations
Research Models Citations
Therapeutics Citations
External Citations
Further Reading
Primary Papers
- Zhao P, Xu Y, Jiang L, Fan X, Li L, Li X, Arase H, Zhao Y, Cao W, Zheng H, Xu H, Tong Q, Zhang N, An Z. A tetravalent TREM2 agonistic antibody reduced amyloid pathology in a mouse model of Alzheimer's disease. Sci Transl Med. 2022 Sep 7;14(661):eabq0095. PubMed.
- Zhao P, Xu Y, Fan X, Li L, Li X, Arase H, Tong Q, Zhang N, An Z. Discovery and engineering of an anti-TREM2 antibody to promote amyloid plaque clearance by microglia in 5XFAD mice. MAbs. 2022 Jan-Dec;14(1):2107971. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Alector
TREM2 modulating antibodies lead a paradigm shift in the therapeutic strategy for Alzheimer’s disease.
For the last two decades, direct targeting of misfolded proteins was the dominant approach for treating neurodegenerative diseases. Over 40 anti-Aβ drugs, 30 and anti-tau drugs, as well as multiple drugs targeting SOD, huntingtin, and α-Synuclein had reached the clinic, largely with disappointing results. We are now witnessing the emergence of immuno-neurology (IN) as a paradigm shift in the therapeutic strategy for neurodegeneration. The goal of IN is to recruit the microglia brain immune system to counteract multiple disease pathologies. IN is conceptionally akin to the immuno-oncology revolution which transformed cancer therapy by recruiting the immune system to destroy tumors. IN is supported by human genetics and by our emerging understanding of the role of microglia in the removal of misfolded proteins, myelin debris and damaged cells, in the recycling and activity of synapses, in neuronal health, and in the function of oligodendrocytes, astrocytes, and endothelial cells.
Human genetic studies of late-onset sporadic Alzheimer’s disease (AD) had surprisingly identified multiple microglia-specific genes and intragenic mutations regulating microglia-expressed genes as key risk factors for AD initiation and possibly progression. For example, familial heterozygous loss-of-function mutations in TREM2, an activating receptor that regulates the survival, proliferation, and function of microglia, were shown to triple the risk for AD. Likewise, familial homozygous loss-of-function mutations in TREM2 or in its signaling co-receptor DAP12/TyroBP invariably lead to Nasu Hakola disease, an early onset form of neurodegeneration with myelin pathology.
Conversely, levels of sTREM2, the TREM2 cleavage product, which likely correlate with higher expression and increased activity of TREM2 in microglia, were shown to be associated with delayed accumulation of Aβ plaques and phospho tau, slowdown in the rate of cognitive decline, slowdown in the rate of brain tissue loss, slowdown in the conversion from mild cognitive decline to AD, and delayed onset and better survival with AD. Consistent with these findings, MS4A, a microglia gene cluster encoding four transmembrane proteins from the CD20 family, was shown to modulate the level of sTREM2/TREM2 and to be protective in AD.
Propelled by human genetics, supportive animal model data and better understanding of microglia biology, we are now seeing the emergence of new therapeutics that target risk genes such as TREM2 and MS4A as levers to recruit microglia and counteract multiple AD disease pathologies. There is currently one TREM2 activating antibody, designated AL002, that is being tested in a double-blind Phase 2 trial for Alzheimer’s disease. In human subjects AL002 was shown to elevate multiple microglia biomarkers including soluble CSFR1, IL1RN and SPP1, indicating target engagement. Other TREM2-activating drugs are currently entering Phase 1 studies in healthy volunteers.
The work by Zhao at al. is another example of a TREM2-activating biologic that combines protein engineering methodologies with cell biology and animal models. Because TREM2 likely requires oligomerization (rather than dimerization) to transduce signal, Zhao at al. constructed a tetravalent antibody that carries four copies of the TREM2-binding domain designated Ab18 TVD-Ig. Ab18 TVD-Ig was up to 100-fold more potent than the parental Ab18 antibody in its ability to activate TREM2 signaling in culture as measured by SYK phosphorylation, 33-fold more potent in phagocytosis of lipidated Aβ, ~300-fold more potent in stimulating migration of microglia toward Aβ, and ~100-fold more potent in promoting the survival of microglia under limited concentration of CSF. Surprisingly, unlike other TREM2 activating antibodies, stimulation of TREM2 by Ab18 TVD-Ig did not change the levels of soluble TREM2, membrane TREM2 or total TREM2, indicating that TREM2 activation does not necessarily induce cleavage or internalization.
Zhao at al. further added a transferrin-binding domain to their tetravalent antibody to generate Ab18 TVD-Ig/aTfR to increase blood-brain barrier (BBB) penetration. Ab18 TVD-Ig/aTfR demonstrated 10- to 30-fold higher concentration of the antibody in the brain compared to the Ab18 TVD-Ig antibody 24 hours after injection and retained 10-fold better CSF-to-serum antibody ratio up to seven days after injection. Consistent with better brain penetration, Ab18 TVD-Ig/aTfR but not the Ab18 TVD-Ig demonstrated potent ability to reduce Aβ plaque burden in 5XFAD mice, with a 10-fold reduction in large Aβ plaques as well as a fivefold increase in clustering of microglia around plaques. Ab18 TVD-Ig/aTfR also considerably decreased the intensity of dystrophic neurons as measured by LAMP1 staining and improved synaptic functions as evidenced by the 40 percent increase in the intensity of the staining of synaptic markers synaptophysin and NeuN. Ab18 TVD-Ig/aTfR further improved anxiety-like behaviors as measured by the elevated plus maze and cognitive function as measured by the contextual and cued fear conditioning test. TVD-Ig/aTfR reduced tau hyperphosphorylation and neurofilament protein phosphorylation by up to 70 percent. The in vivo effects of TVD-Ig/aTfR were largely not observed with Ab18 TVD-Ig, indicating that improved blood barrier penetration was essential for the biological activity of this TREM2-activating construct.
In summary, Zhao at al. have demonstrated potent TREM2 signaling and microglia activation in culture as well as consistent beneficial effect in an AD disease model using a tetravalent TREM2-activating antibody with a transferrin domain. TVD-Ig/aTfR engineered antibody was significantly superior to the corresponding dimeric antibody Ab18 and to the tetravalent antibody lacking the transferrin domain Ab18 TVD-Ig. The findings further validate immuno-neurology and TREM2 activators as exciting approaches to address Alzheimer’s disease and possibly other neurodegenerative disorders.
Given the multiplicity of TREM2 activators that show benefit in animal models, the question arises as to what is the best way to activate TREM2 therapeutically? Specifically, is a tetravalent antibody which carries risks associated with immunogenicity and manufacturability a viable option? The EC50 of the bivalent component of Ab18 TVD-Ig is quite low (79.4 nM), which may necessitate poly-valency to compensate for the low affinity. Likewise, brain-penetration-enhancing technology may carry disadvantages by reducing the half-life of the antibody in the serum, directing it toward red blood cells that express high levels of the transferrin receptor, and possibly recruit tissue macrophages to red blood cells leading to adverse effects.
But again, the low affinity of the TREM2-binding domain may necessitate brain penetration-enhancing technology in this case. High affinity TREM2 antibodies may alleviate the need to generate a tetravalent antibody as well as the need to use brain-penetration-enhancing technology.
Another question is whether excessive TREM2 signaling could lead to overactivation of microglia or to a prolonged period of microglia desensitization. The effects of Ab18 TVD-Ig/aTfR in the 5XFAD model are notable in this regard; however, possible peripheral adverse effects associated with activation of tissue macrophages and engaging red blood cells with a TREM2-activating antibody through the transferring domain were not carefully examined.
With TREM2 and MS4A modulating therapies entering the clinic, we are entering an exciting time in immuno-neurology therapies for AD. We will know soon whether immune modulators of microglia are therapeutically beneficial and whether the simple approach is preferred over the more complex.
Biomedizinisches Centrum (BMC), Biochemie & Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE)
Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE)
Back in 2014 we proposed for the first time that increasing cell-autonomous TREM2 signaling in microglia may be exploited to modulate their protective activities (Kleinberger et al., 2014). In a proof-of-principle study, we inhibited shedding to increase signaling-competent TREM2 on the cell surface. This indeed increased the phagocytic capacity of microglia. Based on these findings we (Schlepckow et al., 2020) and many others developed antibodies (Lewcock et al., 2020), which stimulate TREM2 agonisms (see Table 1). Zhao and colleagues now add yet another antibody to the growing list of TREM2 agonists. However, they now significantly improved agonistic activity by engineering a tetravalent antibody. This antibody dramatically increased syk-signaling (up to 100 times!), Aβ phagocytosis, microglial migration, and survival. These are all phenotypes that are also boosted by many of the previously described agonistic antibodies (see Table 1).
The high degree of consistency of the protective functions mediated by independent agonists is good news for further development of such tools for clinical application.
Unfortunately, this study does not reveal the binding site of the antibody, there is a surprisingly consistent binding of different agonistic antibodies to the stalk region of TREM2 (Table 1), only a few amino acids N-terminal of the ADAM10/17 cleavage site. These antibodies stabilize TREM2 on the cell surface by preventing access of the sheddase to its substrate and by crosslinking TREM2, thereby facilitating cell-autonomous signaling. However, Zhao et al. report that they do not observe a change in soluble TREM2 (sTREM2) nor in total TREM2, which includes the membrane-bound full-length protein. This conclusion is based on western blots, which are difficult to interpret, since the molecular weights of sTREM2 and cellular TREM2 are much lower than usually observed, and they do not discriminate between immature and mature TREM2.
Zhao et al. also increased brain delivery of their antibody by constructing a bispecific antibody using their tetravalent anti-TREM2 antibody and an antibody targeting the mouse transferrin receptor (TfR), which greatly improved brain delivery. Although the aTfR-mediated brain delivery of the antibody works very efficiently, this antibody is cleared more rapidly in the periphery. The authors argue that this could be attributed to broad expression of TfR in peripheral tissues. However, this may raise the concern that using such modified antibodies in patients may cause unwanted side effects in organs with high TfR expression.
Using this modified antibody, 5-month-old 5xFAD mice were treated over 14 weeks with weekly intraperitoneal injections. This led to a massive clustering of microglia around amyloid plaques, increased phagocytosis, and consequently a rather dramatic reduction in plaque number irrespective of their size. But not only that, even neuritic pathology, p-tau, and neurofilament (NFH) were all reduced as well, whereas synapses were maintained, suggesting a rather strong reduction of neurodegeneration and cell death. Strikingly, this was also associated with an improvement of cognition.
Again, similar effects on amyloid pathology were also shown by other antibodies (see Table 2), although not to such a striking extent. The overall consistency of these great findings further strengthens the exciting possibility of therapeutically modulating microglial function. If antibodies could even be further improved, as nicely shown in this study, we may soon have novel and rather unexpected treatments ready for clinical testing.
Although this is great news, we want to point out that agonistic microglia targeting antibodies are not novel wonder drugs. Based on our recent study on CSF sTREM2 in the DIAN cohort, one must assume that, similar to anti-amyloid therapeutic strategies, microglial modulation must also be initiated extremely early during disease development, namely in the seeding phase, which may occur up to 20 years before disease onset. That's the time point when amyloid starts to precipitate in DIAN patients and microglia respond by increasing TREM2 (Morenas-Rodriguez et al., 2022). Simulating the seeding process in mouse models, we were able to show that seeds are readily recognized by microglia and rapidly removed by a TREM2 dependent mechanism (Parhizkar et al., 2019).
One may now also consider dual treatments with anti-amyloid antibodies and microglial agonists. Based on early findings from our lab, this may improve amyloid plaque clearance, as anti-Aβ antibody-mediated plaque reduction is also TREM2-dependent (Xiang et al., 2016).
Table 1
Table 2
References:
Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014 Jul 2;6(243):243ra86. PubMed.
Schlepckow K, Monroe KM, Kleinberger G, Cantuti-Castelvetri L, Parhizkar S, Xia D, Willem M, Werner G, Pettkus N, Brunner B, Sülzen A, Nuscher B, Hampel H, Xiang X, Feederle R, Tahirovic S, Park JI, Prorok R, Mahon C, Liang CC, Shi J, Kim DJ, Sabelström H, Huang F, Di Paolo G, Simons M, Lewcock JW, Haass C. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol Med. 2020 Apr 7;12(4):e11227. Epub 2020 Mar 10 PubMed.
Lewcock JW, Schlepckow K, Di Paolo G, Tahirovic S, Monroe KM, Haass C. Emerging Microglia Biology Defines Novel Therapeutic Approaches for Alzheimer's Disease. Neuron. 2020 Dec 9;108(5):801-821. Epub 2020 Oct 22 PubMed.
Morenas-Rodríguez E, Li Y, Nuscher B, Franzmeier N, Xiong C, Suárez-Calvet M, Fagan AM, Schultz S, Gordon BA, Benzinger TL, Hassenstab J, McDade E, Feederle R, Karch CM, Schlepckow K, Morris JC, Kleinberger G, Nellgard B, Vöglein J, Blennow K, Zetterberg H, Ewers M, Jucker M, Levin J, Bateman RJ, Haass C, Dominantly Inherited Alzheimer Network. Soluble TREM2 in CSF and its association with other biomarkers and cognition in autosomal-dominant Alzheimer's disease: a longitudinal observational study. Lancet Neurol. 2022 Apr;21(4):329-341. PubMed.
Parhizkar S, Arzberger T, Brendel M, Kleinberger G, Deussing M, Focke C, Nuscher B, Xiong M, Ghasemigharagoz A, Katzmarski N, Krasemann S, Lichtenthaler SF, Müller SA, Colombo A, Monasor LS, Tahirovic S, Herms J, Willem M, Pettkus N, Butovsky O, Bartenstein P, Edbauer D, Rominger A, Ertürk A, Grathwohl SA, Neher JJ, Holtzman DM, Meyer-Luehmann M, Haass C. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci. 2019 Feb;22(2):191-204. Epub 2019 Jan 7 PubMed. Correction.
Xiang X, Werner G, Bohrmann B, Liesz A, Mazaheri F, Capell A, Feederle R, Knuesel I, Kleinberger G, Haass C. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol Med. 2016 Sep 1;8(9):992-1004. PubMed.
Washington University School of Medicine
This is an inspiring study. Our group (Wang et al., 2020), among others (Schlepckow et al., 2020; Cheng et al., 2018; Fassler et al., 2021; Price et al., 2020), had previously proposed and studied the possibility of TREM2-based immunotherapy in the AD mouse model. In the current study, the authors further improved the blood-brain permeability of anti-TREM2 antibody (Ab18 TVD-Ig) by fusing to the anti-mouse transferrin receptor (Ab18 TVD-Ig/aTfR), thereby greatly increasing the antibody concentration in the brain parenchyma. Moreover, by engineering Ab18 to a tetravalent form, the authors obtained higher activation of TREM2, such that AD-related pathology in the 5XFAD mouse model was greatly improved after a long-term antibody treatment. The alleviated pathology further ameliorated the behavioral deficits in the AD mouse model.
Studies from different groups have consistently demonstrated the potential and reliability of targeting/activating TREM2 as immunotherapy for AD. This study makes a robust case for Ab18 TVD-Ig as a clinical trial candidate. It will be important to test whether an anti-human TREM2 antibody engineered as Ab18 TVD-Ig is effective in mouse AD models expressing human TREM2 variants, including especially the R47H variant that is considered a high-risk variant.
Interestingly, the authors showed that Ab18 TVD-Ig binding effectively triggers TREM2 clustering without altering TREM2 expression or sTREM2 levels. Since the TREM2 pathway sustains the generation of disease-associated microglia (DAM), one would expect the Ab18 TVD-Ig treatment to increase the expression of TREM2, which is one of the main signature genes of DAM. It would be important to compare how microglia respond to the antibody-mediated activation upon acute and chronic treatment by single-cell RNA sequencing, particularly how treatment impacts DAM, IFN-responsive, and proliferating microglia.
References:
Wang S, Mustafa M, Yuede CM, Salazar SV, Kong P, Long H, Ward M, Siddiqui O, Paul R, Gilfillan S, Ibrahim A, Rhinn H, Tassi I, Rosenthal A, Schwabe T, Colonna M. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer's disease model. J Exp Med. 2020 Sep 7;217(9) PubMed.
Schlepckow K, Monroe KM, Kleinberger G, Cantuti-Castelvetri L, Parhizkar S, Xia D, Willem M, Werner G, Pettkus N, Brunner B, Sülzen A, Nuscher B, Hampel H, Xiang X, Feederle R, Tahirovic S, Park JI, Prorok R, Mahon C, Liang CC, Shi J, Kim DJ, Sabelström H, Huang F, Di Paolo G, Simons M, Lewcock JW, Haass C. Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol Med. 2020 Apr 7;12(4):e11227. Epub 2020 Mar 10 PubMed.
Cheng Q, Danao J, Talreja S, Wen P, Yin J, Sun N, Li CM, Chui D, Tran D, Koirala S, Chen H, Foltz IN, Wang S, Sambashivan S. TREM2-activating antibodies abrogate the negative pleiotropic effects of the Alzheimer's disease variant Trem2 R47H on murine myeloid cell function. J Biol Chem. 2018 Aug 10;293(32):12620-12633. Epub 2018 Mar 29 PubMed.
Fassler M, Rappaport MS, Cuño CB, George J. Engagement of TREM2 by a novel monoclonal antibody induces activation of microglia and improves cognitive function in Alzheimer's disease models. J Neuroinflammation. 2021 Jan 9;18(1):19. PubMed.
Price BR, Sudduth TL, Weekman EM, Johnson S, Hawthorne D, Woolums A, Wilcock DM. Therapeutic Trem2 activation ameliorates amyloid-beta deposition and improves cognition in the 5XFAD model of amyloid deposition. J Neuroinflammation. 2020 Aug 14;17(1):238. PubMed.
TrueBinding
In this interesting paper, the authors increase blood-brain permeability of their anti-TREM2 antibody by fusion to the anti-mouse transferrin receptor. This already has been reported by many groups, like Genentech's. It would be interesting to investigate its effect on peripheral organs.
They also reported effects on receptor signaling and encouraging microglia to migrate toward and phagocytose Aβ oligomers. It will be good to characterize these oligomers.
They used 5-month-old 5xFAD mice, which have a huge Aβ pathology, and treated them over 14 weeks with weekly IP. With such long-term treatment, which may possibly lead to a massive clustering of microglia and also increases phagocytic activity to engulf amyloid plaques, I am wondering about auto-inflammatory side effects
Make a Comment
To make a comment you must login or register.