Protecting Proteasomes from Toxic Tau Keeps Mice Sharp
Quick Links
In neurodegenerative diseases, normal cellular waste disposal systems fail to clear misfolded proteins. One of the systems that goes awry is the proteasome, a meat-grinder-like organelle that chews up unwanted proteins marked with ubiquitin tags. In the December 21 Nature Medicine, researchers led by Karen Duff at Columbia University, New York, in collaboration with Alfred Goldberg at Harvard Medical School, make the case that hyperphosphorylated, aggregated tau directly harms proteasome function, perhaps explaining the failure of this organelle in tauopathies. In tauopathy model mice, stimulating proteasome function by activating protein kinase A cleared accumulated tau and improved learning and memory. Several drugs that activate PKA are already approved by the Food and Drug Administration.
“This is an elegant, meticulous study, and the data look compelling,” said Gerold Schmitt-Ulms at the University of Toronto. Researchers have speculated that proteasome dysfunction might play a key role in tauopathies, but they have struggled to demonstrate this in mouse models, Schmitt-Ulms noted. “The authors show something that many people have suspected, but had trouble documenting. It’s not so straightforward to activate the proteasome in vivo,” he added.
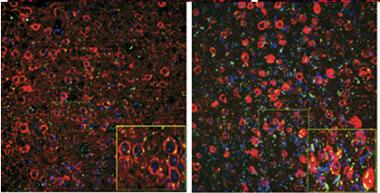
Garbage Pileup.
In five-month-old mice (left) expressing mutant tau (red), cortical neurons largely clear a ubiquitinated protein (green), but by eight months the tagged protein accumulates. [Courtesy of Myeku et al., Nature Medicine.]
Proteasome function slows down in areas of the brains burdened with amyloid pathology (see Keller et al., 2000; Apr 2010 news). Some previous studies have correlated this dysfunction to the presence of hyperphosphorylated, misfolded tau, and found that aggregated tau inhibits proteasomes in vitro (see Keck et al., 2003; Tai et al., 2012). However, these studies stopped short of demonstrating a direct effect in vivo.
To do this, first author Natura Myeku used rTg4510 mice, which express mutant (P301L) human tau and develop tangles starting at about four months of age. She found that proteasome activity in cortical brain extracts from these mice weakened with age, in parallel with worsening tau pathology. The authors crossed rTg4510 mice with a reporter line that expresses a green fluorescent protein labeled for proteasomal destruction (see Lindsten et al., 2003). In the reporter mouse, neurons degraded the GFP quickly and barely fluoresced, but in the P301L tau crosses, green dots accumulated around tau deposits, increasing with age (see image). Moreover, proteasomes from rTg4510 mice co-immunoprecipitated with tau, indicating a physical association. The mutant protein might be obstructing and poisoning proteasomes, the author suggested.
Could normal proteasome function be restored? Elevated levels of cyclic AMP activate protein kinase A, which in turn stimulates proteasome activity by phosphorylating one of its subunits, according to some reports (see Zhang et al., 2007; Metcalfe et al., 2012; Myeku et al., 2012). To test if cAMP could rescue tau toxicity, the authors intraperitoneally injected rolipram, a phosphodiesterase 4 inhibitor that boosts cAMP levels, into three- to four-month-old rTg4510 mice for three weeks. Levels of insoluble and phosphorylated tau dropped by about a quarter, and the animals learned the location of a hidden platform in the Morris water maze as well as wild-type animals, while untreated transgenics performed poorly. Purified proteasomes from treated mice chewed up proteins better than those from untreated mice, and maintained good proteolysis even when exposed to aggregated tau in vitro, suggesting they were protected from its toxic effects.
Although rolipram treatment helped young mice in the earliest stages of tauopathy, for eight- to10-month-old mice the same regimen provided no benefit. These older mice accumulate much more aggregated tau, and the treatment might have to be longer or at a higher dose to have an effect, Myeku suggested. Alternatively, proteasomes from these late-stage mice might resist phosphorylation, she said. In ongoing work, the authors are testing additional pharmacological approaches to raise cAMP levels, some of which might be more potent. They also want to find safer drugs. Rolipram is no longer used in people because of side effects such as nausea, but other, newer drugs in its class, such as apremilast and roflumilast, have better safety profiles, Myeku said. She believes some of these might have therapeutic potential for treating Alzheimer’s disease or other tauopathies.
In addition, some evidence hints that stimulation of proteasomes could provide broad protection against other neurodegenerative diseases. Goldberg recently reported that boosting cAMP in cell lines revved up proteasomes and cleared mutant FUS, SOD1, and TDP-43 in addition to tau (see Lokireddy et al., 2015). “We’re very interested in testing this mechanism of action in mouse models of other neurodegenerative diseases,” Myeku said.
Meanwhile, the details of how tau harms proteasomes remain unclear. The authors speculate that mutant tau may obstruct the access of other proteins to the proteasome, clogging up the machinery. This model implies that mutant tau binds more strongly to proteasomes than does wild-type. However, Schmitt-Ulms recently found the opposite: In a direct comparison, P301L tau bound more weakly to proteasomes than wild-type tau did, leading him to a different interpretation. “We prefer a model whereby the interaction between mutant P301L tau and the proteasome is partially impaired relative to wild-type tau, perhaps contributing to poor proteasome processivity for this substrate. Over time, these altered properties of tau may lead to its aggregation,” he wrote to Alzforum. This aggregated tau may then bind the proteasome and poison it, as Myeku et al. suggest, he added (see full comment below).—Madolyn Bowman Rogers
References
News Citations
Research Models Citations
Mutations Citations
Paper Citations
- Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem. 2000 Jul;75(1):436-9. PubMed.
- Keck S, Nitsch R, Grune T, Ullrich O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer's disease. J Neurochem. 2003 Apr;85(1):115-22. PubMed.
- Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol. 2012 Oct;181(4):1426-35. PubMed.
- Lindsten K, Menéndez-Benito V, Masucci MG, Dantuma NP. A transgenic mouse model of the ubiquitin/proteasome system. Nat Biotechnol. 2003 Aug;21(8):897-902. Epub 2003 Jul 20 PubMed.
- Zhang F, Hu Y, Huang P, Toleman CA, Paterson AJ, Kudlow JE. Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J Biol Chem. 2007 Aug 3;282(31):22460-71. Epub 2007 Jun 12 PubMed.
- Metcalfe MJ, Huang Q, Figueiredo-Pereira ME. Coordination between proteasome impairment and caspase activation leading to TAU pathology: neuroprotection by cAMP. Cell Death Dis. 2012 Jun 21;3:e326. PubMed.
- Myeku N, Wang H, Figueiredo-Pereira ME. cAMP stimulates the ubiquitin/proteasome pathway in rat spinal cord neurons. Neurosci Lett. 2012 Oct 11;527(2):126-31. Epub 2012 Sep 5 PubMed.
- Lokireddy S, Kukushkin NV, Goldberg AL. cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc Natl Acad Sci U S A. 2015 Dec 29;112(52):E7176-85. Epub 2015 Dec 15 PubMed.
Further Reading
News
- Therapeutic Approaches Target Deubiquitinase, Protein Turnover
- Boost APP Ubiquitination, Wnt Signaling to Stave Off AD?
- Garbage BAG2 Takes Out the Tau
- Therapeutic Takedown: Hsp90 Inhibitors Tackle Tau
- Paper Alert: Mass Spec of Tau Points to Ubiquitination
- Ubiquitin System Awry in Ataxia, Intellectual Disability
- Garbage in, Garbage out: Cell Disposal System Linked to Longevity
Primary Papers
- Myeku N, Clelland CL, Emrani S, Kukushkin NV, Yu WH, Goldberg AL, Duff KE. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med. 2016 Jan;22(1):46-53. Epub 2015 Dec 21 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Toronto
The paper by Myeku et al. describes elegant work and documents a critical role of the proteasome in the clearance of mutant tau. The authors are to be complimented for a careful analysis, which makes a compelling case that avenues to boost proteasomal activity might have merit for the treatment of tauopathies. Regarding the mechanism, the authors suggest that mutant tau may poison the proteasome by binding to it, but our recent work on tau protein-protein interactions shows that P301L tau binds more weakly to proteasomes than does wild-type tau (Gunawardana et al., 2015). These data suggest a modified interpretation, consistent with data presented in the new study. (Note that because tau knockout mice, rather than wild-type mice, served as negative controls in co-immunoprecipitations in the Myeku et al. paper, no conclusions about the relative binding propensity of tau versus mutant tau could be drawn.) Thus, we prefer a model whereby the interaction between mutant tau (P301L) and the proteasome is partially impaired relative to wild-type tau, perhaps contributing to poor proteasome processivity for this substrate. Over time, these altered properties of tau may lead to its aggregation and the appearance of proteotoxic effects on the proteasome. In either case, boosting proteasomal activity may compensate for the reduced rate of tau (P301L) clearance.
References:
Gunawardana CG, Mehrabian M, Wang X, Mueller I, Lubambo IB, Jonkman JE, Wang H, Schmitt-Ulms G. The Human Tau Interactome: Binding to the Ribonucleoproteome, and Impaired Binding of the Proline-to-Leucine Mutant at Position 301 (P301L) to Chaperones and the Proteasome. Mol Cell Proteomics. 2015 Nov;14(11):3000-14. Epub 2015 Aug 11 PubMed.
University of Edinburgh
This is an elegant study by Karen Duff's group examining proteasome dysfunction in rTg4510 mice. It is encouraging that enhancing proteasome activity appears to prevent or delay tau-associated pathology and cognitive decline in this model. Since treatment was no longer effective at later disease stages, this implies that for translational benefit, patients might need to be treated very early as is strongly indicated in anti-amyloid therapies. Based on our previous work implicating tau and ubiquitin proteasome system dysfunction in human synapses, it would be interesting to look for synapse recovery in the rolipram-treated mice.
University Maastricht
The ubiquitin-proteasome system #5—Time for a change
The present paper again reports a link between tau-driven 26S proteasome impairment and cognitive dysfunction, here prevented early in disease by activating cAMP-PKA signaling via Rolipram, a phosphodiester-4-inhibitor (PDE 4) developed for depression treatment (for details, see editors’ comments). This collaborative effort between Fred Goldberg and Karen Duff, pioneers in different fields, supports the view that the ubiquitin-proteasome system (UPS) is central in Alzheimer’s disease (AD). They used, as one of the many read-outs, a mouse tauopathy model for frontal temporal dementia with possible implications for AD, crossbred with a mouse reporter model of proteasome dysfunction developed and made available by Nico Dantuma’s group at Karolinska. In the absence of a better model for AD, currently this is the best strategy, while the promising three-dimensional culture system developed for AD by Rudi Tanzi’s group (Choi et al., 2014) may support the conclusions of the present paper. In addition, validation in fixed and frozen postmortem AD tissue may be possible, for instance of the phosphorylation of the Rpn6 proteasome lid subunit.
The present paper is in agreement with the view that the UPS is central in AD, as was already shown in a GWAS study (IGAP, 2015) and a proteomic study (Manavalan et al., 2013). Indeed, from previous evidence it turned out that the UPS is a relevant switch early in AD (e.g., Gentier and Van Leeuwen, 2015). Together, these findings point to proteasome activation via cAMP-PKA signaling, probably via Rpn6 subunit phosphorylation, “a potentially promising therapeutic approach to treatment of proteotoxic diseases,” as the authors write. Their suggestion is in line with recent work (see review of Heckman et al., 2015). In addition, UPS dysfunction resulting in loss of homeostasis may underlie propagation of this toxic tau form, and I suggest the authors include analysis of the brainstem in their mouse model studies. Why? It is now clear that the brainstem may be the area where AD is initiated (Irmler et al., 2012).
References:
Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D'Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, Klee JB, Zhang C, Wainger BJ, Peitz M, Kovacs DM, Woolf CJ, Wagner SL, Tanzi RE, Kim DY. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature. 2014 Nov 13;515(7526):274-8. Epub 2014 Oct 12 PubMed.
International Genomics of Alzheimer's Disease Consortium (IGAP). Convergent genetic and expression data implicate immunity in Alzheimer's disease. Alzheimers Dement. 2015 Jun;11(6):658-71. Epub 2014 Dec 20 PubMed.
Manavalan A, Mishra M, Feng L, Sze SK, Akatsu H, Heese K. Brain site-specific proteome changes in aging-related dementia. Exp Mol Med. 2013;45:e39. PubMed.
Gentier RJ, van Leeuwen FW. Misframed ubiquitin and impaired protein quality control: an early event in Alzheimer's disease. Front Mol Neurosci. 2015;8:47. Epub 2015 Sep 2 PubMed.
Heckman PR, Wouters C, Prickaerts J. Phosphodiesterase inhibitors as a target for cognition enhancement in aging and Alzheimer's disease: a translational overview. Curr Pharm Des. 2015;21(3):317-31. PubMed.
Irmler M, Gentier RJ, Dennissen FJ, Schulz H, Bolle I, Hölter SM, Kallnik M, Cheng JJ, Klingenspor M, Rozman J, Ehrhardt N, Hermes DJ, Gailus-Durner V, Fuchs H, Hrabě de Angelis M, Meyer HE, Hopkins DA, Van Leeuwen FW, Beckers J. Long-term proteasomal inhibition in transgenic mice by UBB(+1) expression results in dysfunction of central respiration control reminiscent of brainstem neuropathology in Alzheimer patients. Acta Neuropathol. 2012 Aug;124(2):187-97. PubMed.
RIKEN Center for Brain Science
Good news for the holiday season. Knock-in substitution of the proteasome subunit phosphorylation site would further confirm the exclusive PKA-proteasome connection because PKA phosphorylates a number of proteins. At the later stage when proteasome activation fails to clear pathological tau, I suspect that autophagy might play some role. In any case, no one would have predicted that proteolysis, considered to be important only for digestion some decades ago, would be attracting so much attention now.
Make a Comment
To make a comment you must login or register.